
Abstract
Background
Neurogenic pulmonary edema (NPE) is a clinical syndrome characterized by the acute onset of pulmonary edema after a significant central nervous system (CNS) insult. NPE occurs as a result of release of catecholamines into the blood immediately after aneurysm rupture. The aim of this study is to investigate the connection between the value of cardiac biomarkers on admission and incidence of NPE in patients with aneurysmal subarachnoid hemorrhage (SAH).
Methods
A total of 262 SAH patients (162 women, 100 men) were prospectively included in the study. Clinical characteristics, electrocardiographic (ECG) changes, serum cardiac and inflammatory biomarkers were measured on admission and on the day of development of NPE. These data were analyzed in order to predict the development NPE.
Results
Nineteen patients (7.25%) developed NPE. Comparison revealed that patients who subsequently developed NPE, sustained more severe SAH. Cardiac damage was more severe in these patients, as represented by significantly higher mean values of all examined cardiac biomarkers (P = 0.000), except for troponin I value that was significantly lower (P = 0.000). Multivariate regression analysis revealed that elevated troponin I (OR, 4.980; 95% CI, 1.27-19.49; P = 0.021) and white blood cell count (OR, 22.195; 95% CI, 3.99-123.50; P = 0.000) are predictors of NPE.
Conclusions
Significantly higher values of cardiac biomarkers were observed in SAH patients complicated with NPE. Elevated values of cardiac biomarkers appear to play an active role in prediction of NPE, although white blood cell count may be involved in the prediction of NPE. There is an influence of SAH therapy on predictors of NPE.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Neurogenic pulmonary edema (NPE) is a clinical syndrome characterized by the acute onset of pulmonary edema after a significant central nervous system (CNS) insult [7]. In patients with subarachnoid hemorrhage (SAH), reports of NPE incidence range from 2 to 42.9% [2, 7, 11, 12, 15–17, 20, 23, 26, 28].
NPE occurs as a consequence of releasing of catecholamines into the systemic circulation immediately after aneurysmal rupture [2, 20, 23, 28].
It is the prevailing view that the autonomic response to elevated intracranial pressure (ICP) plays an important role in the pathogenesis of NPE. Neurological conditions that cause abrupt, rapid and extreme elevation in ICP appear to be at greatest risk of being associated with NPE [9, 19].
Clinically, the likelihood of developing NPE following SAH correlates with increasing age, delay to surgery, vertebral artery origin and the severity of clinical and radiographic presentation (e.g., Hunt-Hess and Fisher grades) [7, 25, 26].
NPE can be associated with cardiac dysfunction although the pathophysiology is considered to be more complex [1, 6, 8, 21, 32].
Since the catecholamines have direct effect on the myocardium, the aim of this study was to investigate the connection between the value of cardiac biomarkers on admission and incidence of NPE in patients with aneurysmal SAH.
Materials and methods
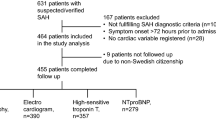
From August 2009 to January 2014, the study enrolled 262 consecutive patients admitted to the Neurosurgery Clinic. Inclusion criteria for the study were patients ≥18 years of age and the diagnosis of aneurysmal SAH confirmed by cerebral computed tomography (CT) scanning and CT angiography [22]. If the CT angiogram was negative and the suspicion of aneurysm existence was high, the CT angiography was repeated 2 days later. The time interval between SAH onset and hospital admission was under 96 h. Chest X-rays were obtained immediately for those suspected of having SAH.
Patients with a history of myocardial infarction, cardiomyopathy or congestive heart failure were excluded from the study.
The study protocol was approved by the Ethics Committee of the School of Medicine, University of Belgrade (No. 440/VI-11), and informed consent was obtained from each patient or an appropriate designee. The identity of the enrolled patients was protected. The followed procedures were in accordance with institutional guidelines.
Demographic and clinical data were collected from patient and family interviews, and by the review of medical records after enrolment. These data included age, sex and risk factors for coronary artery disease (CAD). Heart rate (HR) and systolic blood pressure (SBP) on admission were recorded. The severity of neurological injury was graded using each subject’s admission Hunt and Hess [14] and Fisher grade [10].
Patients with Hunt and Hess grades I and II were admitted to the neurosurgery ward while those with Hunt and Hess grades III-V to the neurosurgical intensive care unit (NICU).
Clinical management
Every patient was examined daily for the occurrence of NPE. Clinical criteria for NPE included the presence of crackles that suggested fluid in the lungs and presence of frothy pink tracheal fluid. Radiographic criteria for NPE included sharply defined pulmonary markings accompanied by blurring or haziness of the perivascular outlines and loss of demarcation of hilar shadows [23]. All chest X-rays were interpreted by board-certified radiologists blinded to the clinical symptoms of the patient. Patients who fulfilled both clinical and radiographic criteria were diagnosed with NPE. Among the 262 SAH patients, 19 (7.25%) developed NPE. In patients with NPE, arterial blood gas analysis was done and the type of respiratory support was determined. The time of occurrence of NPE from SAH attack was noted.
The need for inotropic or vasopressor support and time of initiation were documented.
All NICU patients had continuous electrocardiographic (ECG) monitoring and noninvasive blood pressure monitoring (NIBP), as a part of the standard clinical practice.
Although central venous catheters were placed routinely, pulmonary artery catheters were not placed [11].
In all patients, intravenous nimodipine infusion was started with 5 μg/kg/h and was progressively increased during the following 5 h to a maximum dose of 25 μg/kg/h. This dose was maintained for 21 days if there were no side effects, such as hypotension, which led to decrease or stop of nimodipine intake. In patients with Hunt and Hess grade ≥3, intravenous infusion of 20% solution of mannitol was initiated with first dose of 0.7 g/kg and then 0.35 g/kg every 6–8 h, for 4 days.
Triple-H therapy was initiated in patients with symptomatic vasospasm. However, for patients with NPE, efforts were made to keep them normovolemic instead of hypervolemic to avoid exacerbation.
In case a patient needed continuous sedation in NICU, midazolam (0.1-0.2 mg/kg/h) in combination with remifentanil (0.05-0.2 μg/kg/min) was used. Endotracheal intubation and mechanical ventilation were started when it was clinically indicated.
ECG, serum cardiac and inflammatory biomarkers
Each subject was assessed on the day of enrollment. The subjects with NPE were assessed also on the day they developed NPE.
On each study day, 12-channel ECG was done to every subject in search of ST-T changes (ST depression or elevation, negative, biphasic or flattened T waves), prolonged QTc interval, rhythm disturbances and ventricular premature complexes. All ECGs were interpreted by board-certified cardiologists.
On each study day, the following biomarkers of cardiac injury and inflammatory biomarkers were measured: creatine phosphokinase (CPK), creatine phosphokinase MB isoenzyme (CPK-MB), creatine phosphokinase MB mass concentration (CPK-MB mass), myoglobin, and cardiac troponin I, C-reactive protein (CRP), white blood cell (WBC) count and blood glucose level.
Total serum CPK activity was measured by standard spectrophotometry, as well as the CPK-MB isoenzyme activity. Serum level of troponin I was measured with a chemiluminescence enzyme immunoassay, as well as CPK-MB mass serum level. Serum myoglobin level was measured by chemiluminescence microparticle assay and CRP was measured by turbidimetry. WBC count and glucose levels were measured on standard way.
Troponin I levels, as well as the levels of others biomarkers, were then dichotomized as elevated for levels exceeding next referent values: CPK >150 IU/L in women and 200 IU/L in men was determined as elevated, CPK-MB >24 IU/L was determined as elevated, and CPK-MB mass >5 ng/mL. Troponin I >0.04 ng/mL was determined as elevated and myoglobin >110 ng/mL. CRP >5 mg/L was determined as elevated and WBC count >9.7 × 109/L, as well as blood glucose level >6.1 mmol/L.
The time from SAH symptoms onset to measurement of biomarkers was recorded for every patient and it was less than 96 h.
Neurological assessment
Every patient was neurologically examined in search for signs of hydrocephalus, delayed cerebral ischemia (DCI) or rerupture. In case of neurological deterioration, the CT scan would be done.
Statistical analysis
Two hundred sixty-two patients were dichotomized based on the development of NPE. Data were described by frequency and percentage in case of categorical variables, while mean and standard deviation (SD) were run for continuous variables. The normal distribution of the latter was assessed by Kolmogorov-Smirnov test. For the comparison between NPE+ and NPE– groups, chi-squared or Fisher’s exact test was used for categorical variables, while the unpaired t-test or Mann-Whitney U test was used for continuous variables, where appropriate. Univariate and multivariate logistic regression analysis were used to identify variables predictive of NPE+ outcome. Pearson’s correlation between all potential predictors was examined. SPSS 22 (IBM, Chicago, IL, USA) was used for statistical analysis and P < 0.05 was considered statistically significant.
To minimize the effect of SAH therapy on the results, we have done additional statistical analysis and considered only the patients admitted within the first 24 h of SAH onset for univariate and multivariate logistic regression analysis.
Results
The study included 262 subjects, of which 7.25% (19 subjects) developed NPE. The total number of examined SAH patients was 368. The number of excluded patients was 106 (28.8%). The reasons for exclusion were: 54 patients were admitted at the hospital more than 96 h after SAH onset, 21 patients had a history of myocardial infarction, 16 patients had a history of cardiomyopathy and 15 had a history of congestive heart failure.
As shown in Table 1, the mean age of the 19 study subjects who developed NPE was 52.4 and the 243 who did not develop NPE was 52.2. There were 84.2% of women in patients with NPE and 60.1% in patients without NPE (P = 0.037). History of hypertension (HTA) was more common in patients with NPE (P = 0.02). There were significantly more patients with Hunt and Hess grade >2 (P = 0.005) in patients with NPE, compared to patients without NPE. Comparison of radiological characteristics revealed a lack of any significant difference in occurrence of NPE in patients with Fisher grade >2 (P = 0.067). However, patients with NPE had significantly higher mean Fisher grade than the group without NPE (3.36 ± 0.76 vs 2.86 ± 0.84, P = 0.11). Neither the mean peak SBP on admission nor the heart rate were significantly different in patients with and without NPE. Of those without NPE, 5.35% had atrial fibrillation, while none of those with NPE had this heart rhythm disturbance.
ECG ST-T changes were more common in patients who developed NPE (P = 0.016). Regarding separately, ST depression was more common in subjects with NPE (P = 0.028), as well as negative T waves (P = 0.001) and prolonged QTc interval (P = 0.008).
Cardiac damage was more severe in patients with NPE, as represented by significantly higher mean of all examined cardiac biomarkers (P = 0.000), except troponin I which was significantly lower (P = 0.000). Also, the occurrence of NPE was significantly higher in patients with elevated values of all cardiac biomarkers (P ranged from 0.000 to 0.032). Mean WBC count and elevated WBC count were significantly higher in patients who developed NPE (P = 0.000).
Among neurological characteristics, none of them were statistically significant in patients with NPE comparing to patients without NPE.
The univariate relationships between the predictor variables and NPE, as determined by logistic regression, are shown in Table 2. A Hunt-Hess grade of ≥3, females, history of HTA, hydrocephalus, ST-T changes, prolonged QTc interval were all significant predictors of NPE. Cardiac biomarkers predictive for NPE were: higher values of myoglobin, CPK, CPK-MB fraction and MB mass, WBC count and CRP. Elevated values of cardiac biomarkers were significantly more frequent in patients with NPE (P ranged from 0.000 to 0.017).
Between all the potential predictors examined in univariate logistic regression analysis, we did Pearson’s correlation and found that there was statistically significant correlation between cardiac biomarkers (troponin I, myoglobin, CPK, CPK-MB and CPK-MB mass) and their categorical equivalents (elevated values) (r ranged from 0.158 to 0.836, P from 0.000 to 0.010). Also, we found statistically significant correlation between WBC count, elevated WBC count and CRP (r ranged from 0.316 to 0.816, P = 0.000) and statistically significant correlation between ST-T changes and prolonged QTc (r = 0.611, P = 0.000).
In case that we include all the variables mentioned above in the multiple regression model, there will exist multicollinearity (a phenomenon in which two or more predictor variables are highly correlated), so the coefficient estimates are unstable and difficult to interpret. So we took one of the variables from each group (elevated troponin I, elevated WBC count and ST-T changes) and put them into the multivariate model together with age, female sex and variables which in the univariate model had P < 0.100 (Hunt and Hess grade ≥3, history of HTA and hydrocephalus).
After statistical adjustment by multivariate logistic regression (Table 3), elevated troponin I [odds ratio (OR), 4.862 per quintile; 95% confidence interval (CI), 1.26-18.74; P = 0.022], elevated WBC count (OR, 21.867; 95% CI, 4.02-118.75; P = 0.000), being female (OR, 5,253; 95% CI, 1.14-24.16; P = 0.033), Hunt and Hess grade ≥3 (OR, 12.593; 95% CI, 1.27-124.79; P = 0.030) and presence of HTA (OR, 3.922; 95% CI, 1.07-14.39; P = 0.039), independent predictors of NPE were established.
The number of patients admitted within 24 h after SAH onset who developed NPE is 13 (5% of included patients) and who did not develop NPE is 116 (47.7% of included patients).
The univariate relationships between the predictor variables and NPE, as determined by logistic regression, are shown in Table 4. The Pearson’s correlation between all potential predictors was examined. We found that correlation between cardiac biomarkers and their categorical equivalents (elevated values) ranged from 0.242 to 0.832 (P from 0.000 to 0.006). We also found statistically significant correlation between WBC count and CRP (r = 0.372, P = 0.000), as well as between ST-T changes and prolonged QTc (r = 0.301, P = 0.001). Since the phenomenon of multicollinearity also existed, we took one variable from every group (elevated troponin I, WBC count and ST-T changes) and put them into multivariate model together with age, female sex, Hunt and Hess grade ≥3 and hydrocephalus (P < 0.100).
Multivariate logistic regression analysis (Table 5) revealed that predictors of NPE in patients who did not undergo SAH therapy are not female gender, Hunt and Hess grade ≥3 and history of HTA, but are presence of hydrocephalus (OR, 8.075; 95% CI, 1.55-42.06; P = 0.013), elevated troponin I (OR, 16.182; 95% CI, 2.46-106.21; P = 0.004) and WBC count (OR, 1.418; 95% CI, 1.12-1.79; P = 0.004).
Discussion
The incidence of NPE after SAH in our cohort of 262 patients is 7.25%, which is comparable with that reported in the literature (2-42.9%) [2, 7, 11, 12, 15–17, 20, 23, 26, 28]. ECG abnormalities and myocardial enzyme release after SAH are well-known and described [3, 24, 27], but the relative contribution of cardiac dysfunction in the pathogenesis of NPE has been unclear. The pathophysiology of NPE is multifactorial. There is evidence that in a subset of patients, neurologic insult leads to direct myocardial injury and the development of NPE. Although the cardiac injury after SAH is more severe in patients who developed NPE, measurement of serum cardiac biomarkers with intention to predict occurrence of NPE has rarely been conducted [18, 23, 31].
In our study, none of the patients on admission had NPE. In the study of Inamasu et. al. [18], 11% of the patients were diagnosed with NPE on admission. The finding in our study may be a consequence of initiated diuretic therapy, which 50.8% of our patients underwent, but the important fact is that also in patients admitted within 24 h of SAH onset we did not find NPE on admission.
According to the results of demographic comparison (Table 1), patients with NPE presented significantly more often with clinically severe hemorrhage (Hunt and Hess grades III-V) compared with patients without NPE [17, 23, 24]. All patients with NPE presented with radiologically severe bleeding (Fisher grades III, IV), as expected [23]. Similarly, ECG abnormalities were also more frequent in the group with NPE (Table 1) [23, 24].
Although previous studies showed that ruptured posterior circulation was a risk factor for NPE [17, 23], the location of aneurysm was not associated with NPE in this study. The lack of statistical difference in this study may also be attributable to the pre-hospital death of those patients.
Significantly higher values of cardiac biomarkers levels on admission (except for troponin I) was observed in our SAH patients complicated with NPE, and the result is in concordance with the cardiac role in pathogenesis of NPE [17, 18, 28, 31]. Since cardiac biomarkers were positively correlated, most SAH patients complicated by NPE exhibited a concomitant increase in these cardiac biomarkers levels on admission. Univariate logistic regression analysis revealed that higher levels of cardiac biomarkers on admission could predict NPE, as well as their elevated values.
The serum troponin I level on admission is unexpectedly significantly lower in patients who will develop NPE compared to those who will not, but the elevated values of troponin I were significantly more common in patients who would develop NPE, which is in concordance with previous studies [18, 23, 24, 28]. The important fact in our study is that none of our patients on admission had NPE, so that is probably the reason for such a result.
Also, the authors had serial measurements of troponin I in similar studies [24, 29] and they have chosen the maximal. We had only one measurement, so we believe this may be the source of our different results.
The possibility of prediction of NPE by the elevated WBC is in concordance with studies in which NPE is prevented by attenuating inflammation [4, 5, 13].
Multivariate logistic regression analysis revealed that elevated troponin I and WBC count are predictors of NPE.
For patients admitted within 24 h of SAH onset, cardiac and inflammatory biomarkers with their absolute and elevated values had also the role in prediction of NPE.
According to the ECG changes, the occurrence of NPE is more frequently in patients with these changes, but they are not predictors of NPE.
The influence of SAH therapy on predictors of NPE is obvious. We have found different results in patients admitted within 96 h of SAH onset (female gender, Hunt and Hess grade ≥3, history of HTA) to those admitted within 24 h of SAH onset (presence of hydrocephalus) and in which SAH treatment was not initiated.
There are several limitations of this study. First, we had no possibility to use transthoracic echocardiography (TTE) examination of the heart structure and function. This could enable assessment of the role of the heart dysfunction in pathophysiology of NPE. Zaroff et al. [31] found that the great majority of patients with NPE were complicated by concomitant wall motion abnormality. In another study, Inamasu et al. [17] also found that 88% of the patients with NPE had also Takotsubo cardiomyopathy. Tung et al. [30] revealed that 23% of patients with regional wall motion abnormalities had pulmonary edema.
Secondly, a different time from SAH onset to admission at the hospital could also have influenced the results. Third and finally, we had only one measurement for the examined variables which are measured within 96 h of SAH onset. Of these values, 50.8% were obtained after initiation of SAH therapy and the influence of this therapy on the results is excluded by additional statistical analysis.
Despite these limitations, this study is unique because cardiac biomarkers on admission in prediction of SAH-induced NPE were evaluated for the first time, and we believe that it will provide a new insight into the pathogenesis. The possibility of prediction of NPE occurrence in patients with SAH which may improve outcomes of NPE patients also needs to be further evaluated, as well as the influence of SAH treatment on the results. Since the number of SAH patients complicated by NPE is relatively small, a multi-center study may be required to clarify the issue.
Conclusions
Markedly higher values of serum cardiac biomarkers were observed in SAH patients complicated by NPE. The elevated values of cardiac biomarkers on admission appear to play an active role in prediction of NPE, although absolute and elevated values of WBC count may be involved in prediction of NPE.
Based on results of this study, we recommend taking blood samples for cardiac biomarkers analysis every day in the first days after admission and then according to clinical status and trend of the values of the biomarkers. Although we did not use TTE examination in our study, we recommend it to confirm or exclude global or regional left ventricular systolic dysfunction and to ensure that therapeutic decisions are based on the cardiac status of each patient.
References
Bahloul M, Chaari AN, Kallel H, Khabir A, Ayadi A, Charfeddine F, Hergafi L, Chaari AD, Chelly HE, Ben Hamida C, Rekik N, Bouaziz M (2006) Neurogenic pulmonary edema due to traumatic brain injury: evidence of cardiac dysfunction. Am J Crit Care 15:462–470
Baumann A, Audibert G, McDonnell J, Mertes PM (2007) Neurogenic pulmonary edema. Acta Anaesthesiol Scand 51:447–455
Brouwers PJ, Wijdicks EF, Hasan D, Vermeulen M, Wever EF, Frericks H, van Gijn J (1989) Serial electrocardiographic recording in aneurysmal subarachnoid hemorrhage. Stroke 20:1162–1167
Chen J, Qian C, Duan H, Cao S, Yu X, Li J, Gu C, Yan F, Wang L, Chen G (2015) Melatonin attenuates neurogenic pulmonary edema via the regulation of inflammation and apoptosis after subarachnoid hemorrhage in rats. J Pineal Res 59(4):469–477
Chen S, Zhu Z, Klebe D, Bian H, Krafft PR, Tang J, Zhang J, Zhang JH (2014) Role of P2X purinoceptor 7 in neurogenic pulmonary edema after subarachnoid hemorrhage in rats. PLoS One 12;9(2):e89042. doi:10.1371/journal.pone.0089042, eCollection 2014
Connor RC (1969) Myocardial damage secondary to brain lesions. Am Heart J 78:145–148
Davison DL, Terek M, Chawla LS. Neurogenic pulmonary edema (2012) Critical Care 16:212.
Di Pasquale G, Andreoli A, Lusa AM, Urbinati S, Biancoli S, Cere E, Borgatti ML, Pinelli G (1998) Cardiologic complications of subarachnoid hemorrhage. J Neurosurg Sci 42(Suppl 1):33–36
Ducker TB, Simmons RL (1968) Increased intracranial pressure and pulmonary edema. 2.The hemodynamic response of dogs and monkeys to increased intracranial pressure. J Neurosurg 28:118–123
Fisher CM, Kistler JP, Davis JM (1980) Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 6(1):1–9
Fontes RB, Agular PH, Zanetti MV, Andrade F, Mandel M, Texeira MJ (2003) Acute neurogenic pulmonary edema: case reports and literature review. J Neurosurg Anesthesiol 15:144–150
Friedman JA, Pichelmann MA, Piepgras DG, McIver JI, Toussaint LG 3rd, McClelland RL, Nichols DA, Meyer FB, Atkinson JL, Wijdicks EF (2003) Pulmonary complications of aneurysmal subarachnoid hemorrhage. Neuorsurgery 52:1025–1031
Fujii M, Sherchan P, Soejima Y, Doycheva D, Zhao D, Zhang JH (2016) Cannabinoid receptor type 2 agonist attenuates acute neurogenic pulmonary edema by preventing neutrophil migration after subarachnoid hemorrhage in rats. Acta Neurochir Suppl 121:135–139. doi:10.1007/978-3-319-18497-5_24
Hunt WE, Hess RM (1968) Surgical risk is related to time of intervention in the repair of intracranial aneurysms. J Neurosurg 28:14–20
Inamasu J, Miyatake S, Tomioka H, Suzuki M, Nakatsukasa M, Maeda N, Ito T, Arai K, Komura M, Kase K, Kobayashi K (2009) Subarachnoid hemorrhage as a cause of out-of-hospital cardiac arrest: a prospective computed tomography study. Resuscitation 80:977–980
Inamasu J, Nakamura Y, Saito R, Kuroshima R, Mayanagi K, Ohba S, Ichikizaki K (2002) Normokalemia and hyperglycemia in subarachnoid hemorrhage patients resuscitated from prehospital cardiopulmonary arrest. Resuscitation 54:255–258
Inamasu J, Nakatsukasa M, Mayanagi K, Miyatake S, Sugimoto K, Hayashi T, Kato Y, Hirose Y (2012) Subarachnoid hemorrhage complicated with neurogenic pulmonary edema and takotsubo-like cardiomyopathy. Neurol Med Chir (Tokyo) 52:49–55
Inamasu J, Sugimoto K, Yamada Y, Ganaha T, Ito K, Watabe T, Hayashi T, Kato Y, Ozaki Y, Hirose Y (2012) The role of catecholamines in the pathogenesis of neurogenic pulmonary edema associated with subarachnoid hemorrhage. Acta Neurochir 154:2179–2185
Kosnik EJ, Paul SE, Rossel CW, Sayers MP (1977) Central neurogenic pulmonary edema: with a review of its pathogenesis and treatment. Childs Brain 3:37–47
Macmillan CS, Grant IS, Andrews PJ (2002) Pulmonary and cardiac sequelae of subarachnoid hemorrhage: time for active management? Intensive Care Med 28:1012–1023
Mayer SA, Lin J, Homma S, Solomon RA, Lennihan L, Sherman D, Fink ME, Beckford A, Klebanoff LM (1999) Myocardial injury and left ventricular performance after subarachnoid hemorrhage. Stroke 30:780–786
Menke J, Larsen J, Kallenberg K (2011) Diagnosing cerebral aneurysms by computed tomographic angiography: meta-analysis. Ann Neurol 69:646–654
Muroi C, Keller M, Pangalu A, Fortunati M, Yonekawa Y, Keller E (2008) Neurogenic pulmonary edema in patients with subarachnoid hemorrhage. J Neurosurg Anesthesiol 20:199–192
Naidech AM, Kreiter KT, Janjua N, Ostapkovich ND, Parra A, Commichau C, Fitzsimmons BFM, Connolly ES, Mayer SA (2005) Cardiac troponin elevation, cardiovascular morbidity, and outcome after subarachnoid hemorrhage. Circulation 112:2851–2856
Ochiai H, Yamakawa Y, Kubota E (2001) Deformation of the ventrolateral medulla oblongata by subarachnoid hemorrhage from ruptured vertebral artery aneurysm causes neurogenic pulmonary edema. Neurol Med Chir (Tokyo) 41:529–534
Solenski NJ, Haley EC Jr, Kassell NF, Kongable G, Germanson T, Truskowski L, Torner JC (1995) Medical complications of aneurysmal subarachnoid hemorrhage: a report of the multicenter, cooperative aneurysm study. Participants of the Multicenter Cooperative Aneurysm Study. Crit Care Med 23:1007–1017
Sugimoto K, Inamasu J, Hirose J, Kato Y, Ito K, Iwase M, Sugimoto K, Watanabe E, Takahashi A, Ozaki Y (2012) The role of norepinephrine and estradiol in the pathogenesis of cardiac wall motion abnormality associated with subarachnoid hemorrhage. Stroke 43:1897–1903
Sommargren CE, Zaroff JG, Banki N, Drew BJ (2002) Electrocardiographic repolarization abnormalities in subarachnoid hemorrhage. J Electrocardiol 35(Suppl):257–262
Tanabe M, Crago EA, Suffoletto MS, Hravnak M, Frangiskakis JM, Kassam AB, Horowitz MB, Gorcsan J III (2008) Relation of elevation in cardiac troponin I to clinical severity, cardiac dysfunction, and pulmonary congestion in patients with subarachnoid hemorrhage. Am J Cardiol 102(11):1545–1550
Tung PP, Olmsted E, Kopelnik A, Banki NM, Drew BJ, Ko N, Lawton MT, Smith W, Foster E, Young WL, Zaroff JG (2005) Plasma B-type natriuretic peptide levels are associated with early cardiac dysfunction after subarachnoid hemorrhage. Stroke 36:1567–1571
Zaroff JG, Rordorf GA, Newell JB, Ogilvy CS, Levinson JR (1999) Cardiac outcome in patients with subarachnoid hemorrhage and electrocardiographic abnormalities. Neurosurgery 44:34–39
Zaroff JG, Rordorf GA, Ogilvy CS, Picard MH (2000) Regional patterns of left ventricular systolic dysfunction after subarachnoid hemorrhage: evidence for neurally mediated cardiac injury. J Am Soc Echocardiogr 13:774–779
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflicts of interest.
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent was obtained from all individual participants included in the study.
Additional information
Comments
Nastasovic and co-workers analyze if cardiac biomarkers could predict neurogenic pulmonary edema (NPE) in aneurysmal subarachnoid hemorrhage (SAH). They prospectively included 262 SAH patients and found 19 patients with NPE.
In my opinion, this is an interesting study about an important problem in neurosurgical intensive care. The weaknesses of the study have been seen and adequately discussed by the authors. For me, the most important limitations are that no ultrasound examination of heart function was available and that the influence of SAH therapy, especially triple-H therapy, on the results described remains unclear. These points should be addressed in future studies about this topic.
Marcus Reinges
Giessen, Germany
Rights and permissions
About this article

Cite this article
Nastasovic, T., Milakovic, B., Marinkovic, J.E. et al. Could cardiac biomarkers predict neurogenic pulmonary edema in aneurysmal subarachnoid hemorrhage?. Acta Neurochir 159, 705–712 (2017). https://doi.org/10.1007/s00701-017-3091-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00701-017-3091-6