
Abstract
Background
Recent evidence has demonstrated that rosiglitazone can attenuate cerebral vasospasm following subarachnoid hemorrhage (SAH). Some studies have shown that rosiglitazone can suppress inflammation and immune responses after SAH. However, the precise molecular mechanisms by which cerebral vasospasm is attenuated is not clear.
Methods
In this study, SAH was created using a “double hemorrhage” injection rat model. Rats were randomly divided into three groups and treated with saline (control group), untreated (SAH group), or treated with rosiglitazone. Using immunocytochemistry, hematoxylin and eosin (HE) staining, and measurement of the basilar artery, we investigated the formation of pathologic changes in the basilar artery, measured the expression of caveolin-1 and proliferating cell nuclear antigen (PCNA), and investigated the role of rosiglitazone in vascular smooth muscle cell (VSMC) proliferation in the basilar artery after SAH.
Results
In this study, we observed significant pathologic changes in the basilar artery after experimental SAH. The level of vasospasm gradually increased with time during the 1st week, peaked on day 7, and almost recovered on day 14. After rosiglitazone treatment, the level of vasospasm was significantly attenuated in comparison with the SAH group. Immunocytochemistry staining showed that caveolin-1 expression was significantly increased in the rosiglitazone group, compared with the SAH group. Inversely, the expression of PCNA showed a notable decrease after rosiglitazone treatment.
Conclusions
The results indicate that rosiglitazone can attenuate cerebral vasospasm following SAH. Up-regulation of caveolin-1 by rosiglitazone may be a new molecular mechanism for this response, which is to inhibit proliferation of VSMCs after SAH, and this study may provide a novel insight to prevent delayed cerebral vasospasm (DCVS).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Despite decades of study, subarachnoid hemorrhage (SAH) remains a devastating complication of ruptured intracranial aneurysms that threatens the lives of many patients worldwide [1]. Delayed cerebral vasospasm (DCVS) is a severe complication following SAH, which contributes to mortality and morbidity [2]. Previous experiments in vivo and in vitro have demonstrated that vascular smooth muscle cell (VSMC) proliferation and phenotypic changes play a key role in the DCVS phase [3–5]. Since the underlying signaling modulation of DCVS after SAH remains unclear, it has become increasingly important to understand the underlying signaling pathways that activate the critical effector cells during the multiple phases of this disease.
Early evidence has shown that inflammation and immune responses play important roles in the development of cerebral vasospasm [6–8] and administration of rosiglitazone, a peroxisome proliferator-activated receptor (PPAR) agonist, reduces this inflammatory reaction in vivo [8, 9]. However, suppression of the inflammatory response may be just one of the molecular mechanisms by which rosiglitazone attenuates cerebral vasospasm. Rosiglitazone can up-regulate the expression of caveolae and caveolin-1, both of which play a significant role in regulating the proliferation of VSMC [10–13]. Therefore, this study investigates and assesses the possible mechanisms underlying the role of rosiglitazone in the suppression of VSMC proliferation in the basilar artery after SAH.
Caveolae are 50– to 100-nm flask-shaped invaginations of the plasma membrane which are found in numerous cell types including smooth muscle cells, endothelial cells, type I pneumocytes and adipocytes. Caveolae are biochemically distinct, because they are rich with cholesterol and sphingolipid, and contain large amounts of caveolin proteins. There are three major caveolins, caveolin-1, −2, and −3, which are encoded by distinct genes. Caveolin-1 is the primary caveolae protein, both in terms of quantity and function, and is very important in maintaining the invaginated structure of the caveolae [11]. Recent reports have discovered that caveolae and caveolin-1 play an important role in regulating vascular smooth muscle proliferation, and overexpression of caveolin-1 attenuates vascular myocyte growth [12]. However, the precise role of caveolin-1 as a determinant of cell responses to mitogens is not entirely clear. Previous evidence suggests that through binding of the platelet-derived growth factor (PDGF) receptor to a conserved caveolin scaffolding domain, caveolin-1 can inhibit PDGF receptor activation, thereby reducing p42/p44 mitogen-activated protein kinase (MAPK) activation and smooth muscle cell (SMC) proliferation [14]. Thus far, there are no studies that have examined the possible functional role of caveolin-1 in cerebral VSMC proliferation after experimental SAH in rats.
Materials and methods
Animals and treatments
The study was approved by the Biomedical Ethics Committee of Medical College of Xi’an Jiaotong University, and conformed to the NIH Guide for the Care and Use of Laboratory Animals. A total of 66 male Sprague Dawley rats weighing 300–350 g were purchased from the Laboratory Animal Center of Xi’an Jiaotong University. Animals were housed and fed in a temperature- and humidity-controlled environment with a standardized light–dark cycle (12 h day/night) for 1 week. Rosiglitazone (Cayman Chemical Co., Ann Arbor, MI, USA) was first dissolved in dimethyl sulfoxide ([DMSO] Sigma-Aldrich, St. Louis, MO, USA; vehicle for rosiglitazone) and then diluted with saline (1:4 ratio). Fifty micrograms of rosiglitazone [9] was injected into the cistema magna of each rat 30 min before the injection of autologous blood.
Animals were randomly assigned to three groups: the saline control group (n = 6) which received an intracisternal saline injection, the SAH group (n = 30), which received an injection of DMSO and autologous blood, and the rosiglitazone group (n = 30), which received an injection of rosiglitazone and autologous blood. All three groups received an equal dose of the drugs. SAH was initiated in these rat models by “double hemorrhage” injection where autologous blood was injected twice through the cistema magna. First, male Sprague Dawley rats were anesthetized with an intraperitoneal injection of 10 % chloral hydrate (0.35 ml/kg) and placed in a prone position. The cistema magna was transcutaneously punctured with a no. 8 scalp needle until the flow of clear cerebrospinal fluid. Then, saline (50 µg) for the saline control group, vehicle (50 µg) for the SAH group, and rosiglitazone (50 µg) for the rosiglitazone group was injected into the cistema magna through the needle. Thirty minutes later, fresh autologous arterial non-heparinized blood (0.1 ml/100 g) was slowly injected into the site. Rats in the saline control group were injected with the same dose of normal saline instead of the blood at 37 °C and were subsequently euthanized on day 1. Pressure was applied to the puncture site for 3 min, and the rat was placed in a 30° head-down position for at least 30 min to permit distribution of blood around the basal intracranial arteries. Forty-eight hours after the first injection, the same injections were repeated. On day 1, 3, 5, 7 and 14 after the second injection, rats in the SAH and rosiglitazone groups were euthanized (n = 6) through deep anesthesia and the fixation-perfusion method. Whenever a rat died, it was replaced in the study by another, randomly chosen rat. The basilar arteries were isolated and processed for immunohistochemical and hematoxylin and eosin (HE) staining.
Histopathological analysis and measurement of basilar artery
Rats were deeply anesthetized with an intraperitoneal injection of 10 % chloral hydrate (0.4 ml/kg). Animals were perfused with 250 ml of normal saline followed by 400 ml of 4 % paraformaldehyde under a perfusion pressure of 120 cm H2O. The whole brain with the basilar artery was removed and post-fixed in paraformaldehyde solution, dehydrated via a graded ethanol series, vitrified with dimethyl benzene, embedded with paraffin and sectioned into 10-µm-thick sections using a microtome. Three sections per animal were processed for HE staining. The staining results were observed at 400× magnification; then two blinded investigators without knowledge of the treatment groups, determined the lumen diameter and the wall thicknesses (intimal layer + medium layer) of the blood vessels using a computerized image analysis system (Image-Pro Plus 6.0, Leica, Germany). The lumen diameter was corrected as:
where CD is the corrected diameter, maxD the maximum diameter, and minD the minimum diameter.
Immunohistochemistry study
The brain sections were de-paraffinized in xylene and hydrated in a decreasing gradient of alcohol to distilled water. Endogenous peroxidase activity was blocked with 3 % H2O2 for 5 min, followed by a brief rinse in distilled water and a 15-min wash in phosphate-buffered saline (PBS). Sections were placed in 0.01 mol/l citrate buffer and heated in a microwave oven at 95 °C for 30 min. Sections were cooled at room temperature for 20 min and rinsed in PBS. Non-specific protein binding was blocked by 30 min of incubation in normal goat serum at room temperature, followed by incubation with primary antibodies (rabbit anti-caveolin-1 polyclonal antibody, diluted at the concentration of 1:400; Bioss, Beijing, P. R. China; Rabbit anti-PCNA polyclonal antibody, 1:400 dilution; Bioss, Beijing, P. R. China) for 24 h at 4 °C and a 15-min wash in PBS. Sections were then incubated with goat anti-rabbit IgG-biotin (diluted 1:200) for 30 min, and streptavidin-horseradish peroxidase (diluted 1:100) for 30 min at 37 °C, and sections were washed with PBS for 15 min after each step. Diaminobenzidine was used as the chromogen, and hematoxylin was used as the counterstain. Sections incubated with PBS in the absence of primary antibodies were used as negative controls. Microscopic observation of the immunohistochemically stained sections was performed by an experienced pathologist blinded to the experimental conditions. The immunoreactivity of all of the molecular markers was analyzed using Image-Pro Plus 6.0 software in five microscopic fields (at 400× magnification) throughout identical regions of the basal artery in each section, and the mean optical density (OD) was calculated. Six animals in each group and five sections per animal were chosen for quantitative analysis.
Statistical analysis
The lumen diameter, wall thicknesses and the mean OD value of the immunohistochemically stained sections were determined by two investigators who did not have knowledge of the treatment groups. SPSS 13.0 (SPSS, Chicago, IL, USA) was used for statistical analyses. All data were presented as mean ± SD. Comparisons among multiple groups were performed using ANOVA followed by Tukey’s tests. A P value of less than 0.05 was considered statistically significant.
Results
General observation
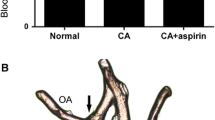
No animals died in the control group, and no obvious neurological deficits were observed. However, a series of neurological symptoms that contained reduced activity and food intake, weight loss, weakened response to stimulation, drowsiness, and even limb paralysis had been discovered in the experimental groups that had SAH. The mortality rate of the animals in the SAH group was 20 % (6 of 30) compared with 16.67 % (5 of 30) in the rosiglitazone treated group. Through craniotomy, we found that animals in the experimental group had excessive blood in their brain, with some blood clots in the basilar cistern and cisterna magna (Fig. 1).

Macroscopic observations of rat brains in each group. No obvious abnormalities were observed in the control group (a); a large amount of blood clots was observed surrounding the arteries in the basilar cistern and cisterna magna after SAH (b)
Measurement of basilar artery
To investigate the vascular changes in the basilar arteries, the mean diameter and thickness of the vessel walls were measured. Representative images of each group are shown in Fig. 2. No obvious pathologic changes were found in the control group. Compared with the control group, significant vasospasm was found in the SAH group and the rosiglitazone group (P < 0.05). However, the level of the vasospasm was markedly attenuated in the rosiglitazone group. Moreover, the mean diameter at each time point was obviously increased (Fig. 3a), and the thickness of the vessel wall was notably declined in the rosiglitazone group (Fig. 3b), compared with the SAH group (P < 0.05). Furthermore, in both the SAH group and the rosiglitazone group, the pathological changes in the basilar arteries became gradually more noticeable, peaked on day 7 and almost recovered on day 14.

Histological changes in the basilar arteries on day 7 after SAH. a–c Representative images of basilar arteries in the control group, in the SAH group and in the rosiglitazone group. No notable abnormality was observed in the control group. Remarkable pathologic changes could be detected in the SAH group, which was attenuated significantly in the rosiglitazone group. e–g Magnified images of the square areas from a through c, respectively. Scale bars 200 and 20 μm, respectively

Representative measurements of the mean diameter and the wall thickness of the basilar arteries. a The mean diameter of the basilar arteries from the control, SAH, and rosiglitazone groups. b The wall thickness of the basilar arteries from each group. The results indicated that, after rosiglitazone treatment, cerebral vasospasm in the basilar arteries was significantly attenuated following SAH. Bars represent the mean ± SD (n = 6, each group). *P < 0.05 compared with the control group, *P < 0.05 compared with the rosiglitazone group
Immunocytochemical study for cavelin-1 and PCNA
We measured the expression of caveolin-1 and PCNA through immunohistochemical analysis. The results showed that cavelin-1 was mainly localized in the membrane of endothelial cells and expressed by SMCs in the basilar arteries. Caveolin-1 was highly expressed by animals in the control group. After SAH, the expression of positive caveolin-1 was significantly attenuated, almost disappeared on day 7 and recovered to normal levels on day 14, compared with the control group. However, rosiglitazone treatment increased caveolin-1 expression at each time point (Fig. 4). PCNA expression was mainly localized to the nucleus of SMCs. PCNA expression was very low in the control group. Inversely, the number of PCNA-positive cells gradually increased after SAH, peaked on day 7, but decreased back to normal levels on day 14. These observations were consistent with the pathologic changes observed in the basilar arteries. In comparison with the SAH group, the PCNA-positive cells showed a significant decline in the rosiglitazone group at each time point, accompanied with the attenuation of pathologic changes (Fig. 5).

Immunohistochemical analysis of caveolin-1 expression in cross sections of the basilar arteries. Caveolin-1 was highly expressed in the vessel walls in the control group (a), almost disappeared in the SAH group (b), and obviously increased in the rosiglitazone group (c). d–f Magnified images of the square area from a through c, respectively. The arrows indicate positively stained cells. Scale bars 200 and 20 μm, respectively. *P < 0.05 compared with the control group, *P < 0.05 compared with the rosiglitazone group

Immunohistochemical analysis of PCNA expression in cross sections of the basilar arteries. PCNA was expressed at a very low level in the vessel walls in the control group (a), increased remarkably in the SAH group b, and obviously declined in the rosiglitazone group (c). d–f Magnified images of the square area from a through c, respectively. The arrows indicate positively stained cells. Scale bars 200 and 20 μm, respectively. *P < 0.05 compared with the control group, *P < 0.05 compared with the rosiglitazone group
Semi-quantitative analysis of the immunohistochemistry data showed that the expression of caveolin-1 was markedly decreased in the basilar arterial wall after SAH, compared with the control group, but after the administration of rosiglitazone the level of caveolin-1 was increased significantly at each time point (P < 0.05). However, PCNA expression was very low level in the control group but elevated gradually after SAH. After rosiglitazone intervention, PCNA expression declined in comparison with the SAH group (P < 0.05). Moreover, correlation analysis between PCNA expression and arterial wall thickness showed a strongly positive correlation (r = 0.91, P < 0.01).
Discussion
Activation of PPAR by its ligand, rosiglitazone, up-regulated caveolin-1 mRNA and protein, and increased the levels of caveolin-1 proteins twofold to fivefold in a concentration-dependent manner within 24 h of treatment [13, 15]. It is presumed that PPAR, as a member of the nuclear hormone receptor superfamily of ligand-activated transcription factors, regulates target gene expression by binding to specific response elements at enhancer sites. In this study, rosiglitazone increased the expression of caveolin-1 in the smooth muscle cells in the basilar arteries. Furthermore, rosiglitazone significantly decreased the thickness of the vessel walls, and decreased the extent of the pathological changes in basilar arteries caused by SAH. On the contrary, rosiglitazone decreased the expression of PCNA in comparison with the SAH group. This study indicates that rosiglitazone inhibits the proliferation of VSMC in the basilar artery in experimental SAH, and upregulates the expression of caveolin-1.
PCNA is a nuclear cyclin and has been found to be the most specific molecular maker for cell proliferation. In the present study PCNA expression increased on day 1 of induction of experimental SAH and peaked on day 7—all in parallel with the changes in vessel wall thickness observed in the HE–stained specimens. Up-regulation of caveolin-1 significantly suppressed the expression of PCNA after rosiglitazone treatment, accompanied by an attenuation in the pathological changes of the vessel wall. The expression of PCNA significantly increased after SAH and implied that VSMCs had switched from a contractile to a synthetic/proliferative phenotype. This has been proven by many studies. Furthermore, the change in the phenotype of VSMC was associated with a loss of plasma membrane caveolae and a decrease in caveolin-1 expression [16].
Caveolae, as a platform for the assembly and compartmentalization of signaling molecules, participates in numerous signaling pathways in both normal and pathological events. Several studies have shown that the close relationship between caveolin-1, growth factor receptor (PDGF) and downstream mediators suggests a functional role for caveolin-1 in the regulation of proliferative events [12, 17, 18]. The present study is the first to describe the role of caveolin-1 in DCVS in the basilar artery in a rat experimental model of SAH. Our observations are highly consistent with previous data in pathological conditions that exhibit hyperproliferative properties or in other cell types, including embryonic fibroblasts, airway smooth muscle cells, and fetal type II epithelial cells from caveolin-1 null mice [17, 19, 20].
There is a direct evidence to suggest that caveolin-1 and caveolae participate in the mechanotransduction and remodeling of blood vessels [21]. Previous reports have demonstrated that caveolin-1 overexpression results in the inhibition of cell-cycle progression and triggers apoptosis in these cells [10]. It was involved in the activation of caspase-3, caspase-9 and other effector caspases [22, 23]. Nevertheless, the underlying signaling pathways and relevant mechanism of how caveolae and caveolin-1 regulate the proliferation of VSMC after SAH is still unknown.
Many studies have demonstrated that PDGF signaling events occur in the specialized plasma membrane microdomain which is known as a caveolae [24, 25] which activates intracellular signal transduction pathways, such as the MAPK pathway, the phosphatidylinositol 3-kinase pathway, and the Ca2+ signal pathway [11, 26–29]. PDGF can be released from blood blots, and PDGF is elevated in the cerebrospinal fluid after SAH. Some studies have shown that PDGF stimulation causes an obvious reduction in the overall caveolae number when compared with untreated VSMCs, consistent with the observation that caveolin-1 levels are reduced under the same conditions. Caveolin-1 is targeted for degradation via the lysosomal pathway after PDGF stimulation and the mechanism for caveolin-1 degradation appears to involve the internalization of cell surface caveolae in response to PDGF stimulation, followed by the merger with the acidic endosomal/lysosomal compartments [12]. Previous results have shown that the proliferative response of VSMCs to PDGF is enhanced in SAH, and that the PDGF-induced proliferation is dependent mainly on early tyrosine phosphorylation [30]. Moreover, phosphorylation of caveolin-1 may be a key step in initiating signal transduction cascades mediated by PDGFR-in human airway smooth muscle cells [17]. However, many direct and/or indirect signaling pathways are involved with the formation of DCVS after SAH and PDGF-induced DCVS may be just one of these underlying pathogenic mechanisms [31]. Subsequent studies should consider the underlying mechanisms of DCVS and the early intracellular signals induced by early brain injury [32, 33].
In conclusion, the results of this study indicate a significant role for rosiglitazone in the suppression of VSMC proliferation in experimental SAH and in the attenuation of the pathological changes in the basilar artery after SAH. Caveolin-1 may play a strong anti-mitogenic role in the inhibition of proliferation of VSMCs after experimental SAH. In the present study, we provide novel data to suggest that it is possible to prevent DCVS after SAH. However, a study of the precise molecular mechanism of this process remains elusive and further experiments are required to address this aspect.
References
Chen S, Feng H, Sherchan P, Klebe D, Zhao G, Sun X, Zhang J, Tang J, Zhang JH (2014) Controversies and evolving new mechanisms in subarachnoid hemorrhage. Prog Neurobiol 115:64–91
Pluta RM (2005) Delayed cerebral vasospasm and nitric oxide: review, new hypothesis, and proposed treatment. Pharmacol Ther 105:23–56
Borel CO, McKee A, Parra A, Haglund MM, Solan A, Prabhakar V, Sheng H, Warner DS, Niklason L (2003) Possible role for vascular cell proliferation in cerebral vasospasm after subarachnoid hemorrhage. Stroke 34:427–433
Miller CA, Lombard FW, Wu C-T, Hubbard CJ, Silbajoris L, Borel CO, Niklason LE (2006) Role of vascular mitogens in subarachnoid hemorrhage-associated cerebral vasculopathy. Neurocrit Care 5:215–221
Suzuki H, Hasegawa Y, Kanamaru K, Zhang JH (2011) Mitogen-activated protein kinases in cerebral vasospasm after subarachnoid hemorrhage: a review. Acta Neurochir Suppl 110:133–139
Crowley RW, Medel R, Kassell NF, Dumont AS (2008) New insights into the causes and therapy of cerebral vasospasm following subarachnoid hemorrhage. Drug Discov Today 13:254–260
Dhar R, Diringer MN (2008) The burden of the systemic inflammatory response predicts vasospasm and outcome after subarachnoid hemorrhage. Neurocrit Care 8:404–412
Wu Y, Tang K, Huang RQ, Zhuang Z, Cheng HL, Yin HX, Shi JX (2011) Therapeutic potential of peroxisome proliferator-activated receptor gamma agonist rosiglitazone in cerebral vasospasm after a rat experimental subarachnoid hemorrhage model. J Neurol Sci 305:85–91
Morgenweck J, Abdel-Aleem O, McNamara K, Donahue R, Badr M, Taylor B (2010) Activation of peroxisome proliferator-activated receptor γ in brain inhibits inflammatory pain, dorsal horn expression of Fos, and local edema. Neuropharmacology 58:337–345
Galbiati F, Liu J, Capozza F, Frank PG, Zhu L, Pestell RG, Lisanti MP (2001) Caveolin-1 expression negatively regulates cell cycle progression by inducing G0/G1 arrest via a p53/p21WAF1/Cip1-dependent mechanism. Mol Biol Cell 12:2229–2244
Parton RG, del Pozo MA (2013) Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol 14:98–112
Peterson TE, Guicciardi ME, Gulati R, Kleppe LS, Mueske CS, Mookadam M, Sowa G, Gores GJ, Sessa WC, Simari RD (2003) Caveolin-1 can regulate vascular smooth muscle cell fate by switching platelet-derived growth factor signaling from a proliferative to an apoptotic pathway. Arterioscler Thromb Vasc Biol 23:1521–1527
Tencer L, Burgermeister E, Ebert MP, Liscovitch M (2008) Rosiglitazone induces caveolin-1 by PPARγ-dependent and PPRE-independent mechanisms: the role of EGF receptor signaling and its effect on cancer cell drug resistance. Anticancer Res 28:895–906
Yamamoto M, Toya Y, Jensen RA, Ishikawa Y (1999) Caveolin is an inhibitor of platelet-derived growth factor receptor signaling. Exp Cell Res 247:380–388
Burgermeister E, Tencer L, Liscovitch M (2003) Peroxisome proliferator-activated receptor-gamma upregulates caveolin-1 and caveolin-2 expression in human carcinoma cells. Oncogene 22:3888–3900
Thyberg J (2000) Differences in caveolae dynamics in vascular smooth muscle cells of different phenotypes. Lab Invest 80:915–929
Gosens R, Stelmack GL, Dueck G, McNeill KD, Yamasaki A, Gerthoffer WT, Unruh H, Gounni AS, Zaagsma J, Halayko AJ (2006) Role of caveolin-1 in p42/p44 MAP kinase activation and proliferation of human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 291:L523–L534
Stehr M, Adam RM, Khoury J, Zhuang L, Solomon KR, Peters CA, Freeman MR (2003) Platelet derived growth factor-Bb is a potent mitogen for rat ureteral and human bladder smooth muscle cells: dependence in lipid rafts for cell signaling. J Urol 169:1165–1170
Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP (2001) Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 276:38121–38138
Wang Y, Maciejewski BS, Drouillard D, Santos M, Hokenson MA, Hawwa RL, Huang Z, Sanchez-Esteban J (2010) A role for caveolin-1 in mechanotransduction of fetal type II epithelial cells. Am J Physiol Lung Cell Mol Physiol 298:L775–783
Yu J, Bergaya S, Murata T, Alp IF, Bauer MP, Lin MI, Drab M, Kurzchalia TV, Stan RV, Sessa WC (2006) Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J Clin Invest 116:1284–1291
Bennett MR (2002) Apoptosis in the cardiovascular system. Heart 87:480–487
Clarke M, Bennett M, Littlewood T (2007) Cell death in the cardiovascular system. Heart 93:659–664
Boucher P, Liu P, Gotthardt M, Hiesberger T, Anderson RG, Herz J (2002) Platelet-derived growth factor mediates tyrosine phosphorylation of the cytoplasmic domain of the low Density lipoprotein receptor-related protein in caveolae. J Biol Chem 277:15507–15513
Liu P, Ying Y, Anderson RG (1997) Platelet-derived growth factor activates mitogen-activated protein kinase in isolated caveolae. Proc Natl Acad Sci U S A 94:13666–13670
Adam RM, Roth JA, H-l C, Rice DC, Khoury J, Bauer SB, Peters CA, Freeman MR (2003) Signaling through PI3K/Akt mediates stretch and PDGF-BB-dependent DNA synthesis in bladder smooth muscle cells. J Urol 169:2388–2393
Smythe GM, Rando TA (2006) Altered caveolin-3 expression disrupts PI (3) kinase signaling leading to death of cultured muscle cells. Exp Cell Res 312:2816–2825
Stary CM, Tsutsumi YM, Patel PM, Head BP, Patel HH, Roth DM (2012) Caveolins: targeting pro-survival signaling in the heart and brain. Front Physiol 3:393
Whitmarsh AJ (2013) A new regulator of caveolae signalling. ELife 2:e01428
Maeda Y, Hirano K, Hirano M, Kikkawa Y, Kameda K, Sasaki T, Kanaide H (2009) Enhanced contractile response of the basilar artery to platelet-derived growth factor in subarachnoid hemorrhage. Stroke 40:591–596
Kolias AG, Sen J, Belli A (2009) Pathogenesis of cerebral vasospasm following aneurysmal subarachnoid hemorrhage: putative mechanisms and novel approaches. J Neurosci Res 87:1–11
Cahill WJ, Calvert JH, Zhang JH (2006) Mechanisms of early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab 26:1341–1353
Kusaka G, Ishikawa M, Nanda A, Granger DN, Zhang JH (2004) Signaling pathways for early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab 24:916–925
Acknowledgments
This study was supported by the National Natural Science Foundation of China, No. 30,471,774; the New Century Excellent Talent Support Project of Ministry of Education, No. NCET-05-0,831.
Author contributions: Jin-Ning Song and Mao-Feng Cheng designed the research; Mao-Feng Cheng and Dan-Dong Li performed the research. Mao-Feng Cheng wrote the manuscript. Yong-Lin Zhao, Ji-Yang An, Peng Sun, and Xian-Hua Luo participated in the study.
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Mao-Feng Cheng and Jin-Ning Song contributed equally to this study.
Rights and permissions
About this article

Cite this article
Cheng, MF., Song, JN., Li, DD. et al. The role of rosiglitazone in the proliferation of vascular smooth muscle cells after experimental subarachnoid hemorrhage. Acta Neurochir 156, 2103–2109 (2014). https://doi.org/10.1007/s00701-014-2196-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00701-014-2196-4