
Abstract
Background
This is a retrospective study of 11 patients harboring a solitary fibrous tumor (SFT) of the central nervous system (CNS), with special emphasis on unusual clinicopathological and outcomes patterns.
Method
Between 2000 and 2008, 11 patients harboring CNS SFTs were treated at our institution. Patient charts were retrospectively reviewed and tumor location, clinical presentation, imaging characteristics, extent of resection, dural origin, pathological features, adjuvant treatment, and follow-up data were collected, focusing on five atypical cases (four intracranial and one within the spine).
Findings
One intracranial SFT arose from the sella turcica and relapsed threefold during the 6 years following partial removal. Disease progressed as successive isolated local recurrences treated by subsequent surgical interventions and gamma-knife radiosurgery. The MiB-1 labeling index analysis showed a steady increase in these sequential recurrences (ranging from less than 3% up to 6%) without obvious malignant transformation. The second SFT occurred in the cerebellopontine angle and exhibited a high MiB-1 index (10%) without noticeable features of malignancy. It relapsed twice during the 5 years following gross total resection without demonstrating a more aggressive histological pattern. The third SFT arose from the cerebellar tentorium, widely invaded the lateral sinus and adjacent bone, had a low MiB-1 index, and has not recurred within the 2 years after incomplete resection. The two remaining SFTs presented with unusual clinicoradiological features. We described a extremely rare case of intraventricular SFT, and a case of extradural SFT of the thoracic spine (T8–T9) radiologically consistent with a schwannoma. Immunohistochemistry confirmed that all tumors were SFTs.
Conclusions
These atypical presentations gave us the opportunity to provide further information about the variability of the clinicoradiological patterns and natural histological course of CNS SFTs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The new World Health Organization classification of tumors of the central nervous system (CNS) [18] includes solitary fibrous tumors (SFTs) among the mesenchymal non-meningothelial tumors. SFTs of the meninges were first described by Carneiro et al. in 1996 [5]. The identification was based on distinctive features common to soft tissue SFTs, including immunoreactivity for CD34 and vimentin but not for epithelial membrane antigen (EMA) or S100 protein (PS100). This immunohistochemical profile differs from that encountered in other more common and better recognized CNS spindle-cell tumors such as fibrous meningiomas or hemangiopericytomas (HPCs) [5, 26]. To date, fewer than a hundred cases of SFTs have been reported in the CNS, generally in short case reports or small series. Because these lesions are relatively rare, diagnosis can sometimes remain difficult, especially if the clinicoradiological presentation is not meaningful. Moreover, although CNS SFTs have generally been described as slow-growing non-aggressive tumors, more information are needed about their natural histological course and biological potential. In this study, we present 11 additional cases of SFT with special emphasis on five cases with atypical features and outcomes, including description of unusual clinicoradiological patterns and histological and MiB-1 index analysis in sequential recurrences.
Methods and materials
Between 2000 and 2008, 11 patients harboring CNS SFTs were treated at our institution. Patient charts were retrospectively reviewed and tumor location, clinical presentation, imaging characteristics, extent of resection, dural origin, pathological features, adjuvant treatment, and follow-up data were collected. In this report, we present our series with special emphasis on five atypical cases with unusual features and outcomes.
All of the surgical specimens were fixed in 10% buffered formalin and embedded in paraffin. Histological sections were stained with hematoxylin–eosin with or without saffron counterstaining, periodic acid-Schiff, and reticulin. Mitoses were counted by examining ten high-power fields (HPF), three times for each tumor, and determining an average number per ten HPF. Immunohistocemichal studies were performed using the standard labeled streptavidin-biotin technique or EnVision peroxidase method (Dako S.A., Trappes, France). Immunohistochemical staining was conducted for CD34, vimentin, PS100, EMA, and Ki-67.
Results
Whole population characteristics, treatment considerations, and survival analysis
Demographic data, clinical presentation, tumor location, and treatment characteristics are summarized in Table 1. There were nine women (81.2%) and two men (18.8%). At the time of diagnosis, the patient ages ranged from 39 to 66 years (median age = 52.3 ± 8 years). The location was supratentorial in five cases (45.5%), infratentorial in five cases (45.5%) and spinal in one case (9%). Signs and symptoms depended on tumor location. At the supratentorial level, intracranial hypertension, slowly progressive neurological deficits, or epilepsia were the most frequent symptoms. At the infratentorial level, cerebellar ataxia or vestibular syndrome were usually found. The patient with the spinal lesion presented with back pain and intercostal neuralgia. On MR imaging, the SFT was isointense with normal cerebral parenchyma on T1-weighted images, iso- to hyperintense on T2-weighted images, and showed intense and homogeneous enhancement after intravenous administration of gadolinium (Figs. 1, 2, and 3). The tumor presented a dural implantation in eight cases (72.7%). Gross total resection was achieved in nine cases (81.8%) whereas subtotal resection was achieved in two cases (18.2%). Gamma-knife radiosurgery was performed at the time of recurrence in two patients (patients 1 and 2) on the basis of a high Ki-67 index. The median follow-up period was 51.5 months (range, 24–78 months). Tumor recurrence or progression occurred in three patients (27.3%). The median delay between initial surgery and recurrent or progressive disease was 23.7 ± 8 .5 months. Progression-free survival curve is shown in Fig. 4.

a, b, and c, typical radiological presentations (dura-based masses) of three intracranial SFTs. Post-contrast T1-weighted coronal (a), saggital (b), and axial (c) MRI scans of SFTs of the cerebellar tentorium (a), foramen magnum (b), and cerebellopontine angle (c). Intravenous gadolinium injection resulted in a strong (and heterogeneous, case c) enhancement of all tumors

Atypical presentation of an intracranial SFT (patient 4). a to c, axial magnetic resonance images of the brain at the level of the atrium of the lateral ventricles. The tumor is a well circumscribed and lobulated left ventricular mass, isointense on T1- (a) and T2-weighted (b) images compared with adjacent brain parenchyma. T1-weighted image with intravenous administration of gadolinium (c) shows a strong and homogeneous enhancement of the lesion

Atypical presentation of an intraspinal SFT (patient 5). Preoperative MRI of the thoracic spine showing a lesion consistent with a schwannoma at T8–T9. a Sagittal, T1-weighted image showing a isointense mass behind T8–T9 vertebral bodies. b Sagittal, T2-weighted image showing hypointense mass. c and d Axial T1-weighted-gadolinium-enhanced image demonstrating a homogeneously enhanced mass, located mainly in the extradural space with foraminal extension and partly in the intradural extramedullary space

Graph of Kaplan–Meier. Survival without progression curve
Detailed description of atypical cases
Patient 1
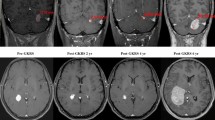
(This case has been previously published [22] but we present here new information relative to the clinical and natural histological course of the tumor). A 66-year-old woman presented in 2000 with progressive amaurosis and visual field impairment. Magnetic resonance imaging (MRI) revealed a lesion in the sella turcica and the suprasellar region radiologically consistent with a pituitary adenoma. The tumor was incompletely removed via a transphenoidal approach and recurred threefold in the same location within 6 years (2002, 2003, 2006), with extension to the optic chiasm, cavernous sinus, and carotid arteries. All recurrences were associated with visual impairment and justified iterative surgical removal via a frontopterional approach. Gamma-knife radiosurgery was performed at the time of the second recurrence (2003) as an adjuvant treatment (marginal dose of 14 Gy). However, the patient died in 2007 of uncontrolled disease progression.
Patient 2
A 39-year-old woman presented in 2004 with a 6-month history of headaches. MRI showed a contrast enhancing ovoid mass in the left cerebellopontine angle measuring 2.5 × 2.5 × 1.5 cm. The tumor was totally removed through a left suboccipital approach. Two years later, the symptoms recurred and MRI demonstrated tumor recurrence in the same location. A repeat craniotomy was performed with incomplete removal because of the invasive nature of the tumor regarding cranial nerves and medulla oblongata. Gamma-knife radiosurgery was performed as an adjuvant treatment at the time of the first recurrence and 3 years after for focal progressive disease (marginal dose of 14 Gy in each procedure). The patient is alive with residual disease.
Patient 3
This 45-year-old woman was admitted for a first epileptic seizure. Computed tomography revealed a homogeneous right temporal supratentorial 3 × 3 × 2.5-cm mass radiologically consistent with a meningioma. MRI demonstrated that the lesion arose from the cerebellar tentorium, extended into the supra- and infratentorial spaces, and invaded the transverse sinus. Operative findings confirmed that the mass widely invaded the transverse sinus but also adjacent bone. The lesion was subtotally removed. Nonetheless, at last follow-up (2 years post-surgery) the patient is asymptomatic without radiological progression of the residual tumor.
Patient 4
A 60-year-old woman presented with a 1-month history of gradually progressive speech disturbances and headaches. The neurological examination at admission revealed Wernicke aphasia with neologisms and moderate weakness of the right side. MRI revealed a well-circumscribed lobulated mass in the atrium of the left ventricle, homogeneously enhanced after intravenous administration of gadolinium, and measuring 5 cm in its largest diameter (Fig. 2). Intraventricular meningioma was considered as the most likely diagnosis preoperatively. Total removal of the tumor was achieved via a left occipitoparietal craniotomy. The tumor was well circumscribed, firm, and fibrous. Inferiorly, the tumor was attached to the choroid plexus. The patient made an uneventful recovery and is disease free 2 years post-surgery.
Patient 5
A 52-year-old woman was admitted with a 6-month history of back pain and left intercostal neuralgia. MRI of the thoracic spine (T8–T9) demonstrated an extradural and partly intradural lesion homogeneously enhanced by gadolinium (Fig. 3). At this time, schwannoma was suggested as the diagnosis, and a hemi-laminectomy, and resection of the tumor was performed. Intraoperatively, the tumor was predominantly extradural, well demarcated, and loosely attached to the dura-mater. After resection of the extradural part of the tumor, a T-shaped incision of the dura and nerve root was performed, and a small piece of the intradural portion of the tumor adherent to the T8 anterior and posterior rootlets was also successfully resected. The patient is actually disease free 4 years post-surgery.
Pathological findings
All of the patients fulfilled the following pathological criteria: presence of spindle-shaped cells arranged in fascicles that alternate with pauci-cellular collagen-rich matrix (Fig. 5a), diffuse immunoreactivity with antibodies against CD34 and vimentin (Fig. 5b), and negative immunoreactivity with antibodies against PS100 and EMA. No myxoid changes were noted except in the primary specimen of sellar SFT (patient 1). None of tumors exhibited nuclear pleomorphism, atypias, or necrosis, that have been associated with malignant SFTs. Mitotic figures were absent in eight cases, scarce in one case (patient 3), and scattered in the cerebellopontine angle SFT (patient 2, first surgical specimen and recurrence 1) and in the sequential recurrences of the sellar SFT (patient 1). In patient 1 sellar SFT, the increased proliferative activity in the sequential recurrences roughly correlated with the MiB-1 labeling index, ranging from less than 3% up to 6%. The MiB-1 labeling index was 10% in the two specimens of patient 2 cerebellopontine angle SFT (first occurrence and recurrence 1), and below 2% (range, 0.5–2%) in the other patients. A focal brain invasion was noted at the time of the first recurrence of the sellar SFT (patient 1).

Pathological findings. a Photomicrographs showing paraffin-embedded tumor sections. The highly cellular tumor is composed of spindle cells arranged in fascicles with intercellular collagen deposition and thin-walled blood vessels (hematoxylin and eosin; original magnification ×200). b Immunohistochemical stains demonstrating strong and diffuse positivity of the tumor cells to CD34 (original magnification ×400)
Discussion
CNS SFTs are recently recognized distinctive dural-based neoplasms of the CNS [3, 10]. They are considered as counterparts of similar lesions initially identified in the pleura by Klemperer and Rabin in 1931 [14], which subsequently have been reported in numerous extrapleural sites [4, 9, 15, 29]. In the CNS, they are known to arise from the meningeal coverings [3, 10] without meningothelial differentiation and are classified as mesenchymal non-meningothelial tumors. Current studies suggest they are derived from vascular mesenchymal cells [8]. At surgery, we found that the intraventricular SFT (patient 3) was attached to the choroid plexus, which is consistent with the former theory. Since their recognition in the CNS [5], fewer than a hundred cases of SFTs have been recorded in the literature. Nevertheless, it is probable that the real frequency of these tumors has been underestimated when the definition of immunohistochemical features of SFTs was not complete, as suggested in the literature by a number of cases of recurrent SFTs with previously erroneous histological diagnosis [25, 26]. In the literature, most SFTs occur in adulthood. The reported median age at the time of diagnosis is approximately 50 years [27] and the sex ratio is 1:1. A few cases have been described in children [16]. Patients with intracranial SFT generally present with slowly progressive neurological deficit or raised intracranial pressure. Back pain and radiculalgia often occur in intraspinal SFTs [1, 2, 28]. It appears that CNS SFTs show a tendency to arise predominantly in the posterior fossa and spine [19]. Intracranially, SFTs typically demonstrate a clearly dural origin and are usually mistaken for meningioma either radiologically and surgically. Intracranial lesions referred to as an intra-parenchymal non-dural-based SFT are extremely rare and have been noted in a handful of cases in the literature [19, 27]. To our knowledge, we presented the fourth reported case of intraventricular SFT [17, 24, 26] and the second case of SFT occurring in the pituitary fossa [6]. Conversely, spinal SFTs usually occur in an intramedullary position, without dural attachment, and cases presenting as an extradural lesion as in our patient 5 seem to be exceptional [13].
Although MRI has proved the modality of choice for detecting, characterizing, and staging most soft tissue tumors, a consensus MRI study regarding CNS SFTs is not yet available. In addition, this entity is still poorly recognized among the imaging community. As a consequence, most SFTs are still radiologically misdiagnosed and/or mistaken for meningiomas or schwannomas as it was observed in our study. With increased recognition of SFTs, radiological studies are reporting imaging features for SFTs [16, 17, 19]. On MR imaging, SFT is usually isointense with normal cerebral parenchyma on T1-weighted images, iso- to hyperintense on T2-weighted images, and shows intense and homogeneous enhancement after intravenous administration of gadolinium. Moreover, it is of interest to note that the typical “dural tail” sign classically seen in meningiomas is usually not present [19].
Similar to the findings in our patients, SFT consists of monomorphic spindle-shaped cells arranged in fascicles and set in a rich collagenous background [12]. Blood vessels are thin-walled and can sometimes suggest the so-called “staghorn vessels” resembling those of HPC [12]. Myxoid changes have been reported in SFTs, particularly in the spinal cord lesions, but pure myxoid variants are extremely rare [1, 2]. Hyperchromatism, nuclear pleomorphism, mitotic figures, and tumor necrosis have been infrequently documented in the so-called malignant variants [7]. The unifying characteristic of all SFTs remains the strong reactivity of the spindle cells for CD34 and vimentin plus their negative staining for PS100 and EMA [8]. The most notable differential diagnosis of intracranial SFT includes fibroblastic meningioma and meningeal HPC [23, 25]. The differential diagnosis is greatly helped by the immunohistochemical findings. Fibrous meningiomas characteristically express EMA and PS100 [23]. HPCs consistently do not stain for EMA and PS100. CD34 expression is observed in 33% of HPCs but staining is focal or tends to be patchy and weak [23]. Nevertheless, based on the absence of pericytic differentiation in most HPCs and the morphological and immunohistochemical similarities to SFTs, the concept that HPC is a variant of SFT is widely accepted [11]. The atypical presentations reported in our study demonstrate the difficulty in diagnosis and emphasize the need for a wider use of immunohistochemical screening, the only method that should enable analysis of the real incidence of CNS SFTs, especially when clinical or radiological features are not meaningful (patients 4 and 5). In this respect, a retrospective analysis with CD34 staining of tumors previously diagnosed as meningioma or HPC might also lead to interesting findings [26].
The treatment of choice is surgical resection. Local recurrences have been reported with incomplete surgical excision [19]. Some cases with malignant potential have also been reported, as well as metastases [7, 19]. Miyashita et al. [20] described a recurrent intracranial SFT with cerebrospinal fluid dissemination in a patient who had a high MiB-1 count. Their case report suggested that SFTs may sometimes transform into a more aggressive histological pattern. Similar to their experience, our patient 1 experienced several recurrences after partial removal. We observed a steady increase of the MiB-1 labeling index with each occurrence, suggesting a worse prognosis and a potential malignant transformation risk. Cerebral microinvasion was noted in the second occurrence, but this finding has not generally been regarded as a sign of malignant transformation but as an expression of increased local aggressiveness [7]. Conversely, our patient 2 cerebellopontine angle SFT exhibited a high proliferative activity from the first occurrence but the sequential histological and MiB-1 index analysis failed to demonstrate a more aggressive histological behavior when it relapsed 2 years later, suggesting the variability of the natural course of CNS SFTs. To date, no study has defined histological criteria for malignant CNS SFTs, and predictors of long-term outcome have not been well defined. In the literature, the invasive nature for cerebral parenchyma, vascular structures, or bone of some SFTs do not necessarily seem to be predictive of malignant behavior, but is described as pejorative for prognosis, with an increasing risk of recurrence if the lesion is not amenable to complete excision [7, 19]. Our patient's lesion (patient 3) widely invaded adjacent structures (bone and transverse sinus) but had a very low MiB-1 index, without noticeable features of malignity, and has not recurred within the 2 years post-surgery suggesting the unpredictable nature and clinical course of CNS SFTs. Although CNS SFTs have generally been described as slow-growing lesions with low recurrence rate after radical surgical excision, the lack of larger series with significant follow-up had failed to establish the long-term biologic behavior of these tumors. Recently, Metellus et al. [19] reported a series of 18 cases of CNS SFTs with a high recurrence or progression rate (50% with a median follow-up period of 45.3 months) suggesting that SFTs may recur even after a long respite period. Although radiosurgery and conformational radiotherapy have been proposed in partially resected or histologically atypical intracranial SFTs [19, 21], as in our patients 1 and 2, it is still difficult to draw definite conclusions regarding the role of adjuvant therapy because very few cases have been described. Therefore, larger series with significant follow-up are needed to allow definite conclusions regarding treatment recommendations and prognosis.
Conclusions
In summary, these atypical presentations demonstrate the difficulty in diagnosis when clinicoradiological features are not meaningful, and the need for a wider use of immunohistochemical screening, the only method that should enable analysis of the real incidence of CNS SFTs. It also further shows the unpredictable nature and variability of the natural course of CNS SFTs, stressing the need to best defined histological predictors of outcome. Meanwhile, careful follow-up remains mandatory for all CNS SFTs.
Abbreviations
- CNS:
-
Central nervous system
- CPA:
-
Cerebellopontine angle
- CT:
-
Computed tomography
- EMA:
-
Epithelial membrane antigen
- GKRS:
-
Gamma-knife radiosurgey
- GTR:
-
Gross total removal
- HPC:
-
Hemangiopericytoma
- HPF:
-
High-power fields
- ICH:
-
Intracranial hypertension
- MRI:
-
Magnetic resonance imaging
- PS100:
-
S100 protein
- SFT:
-
Solitary fibrous tumor
- STR:
-
Subtotal removal
References
Alston SR, Francel PC, Jane JA Jr (1997) Solitary fibrous tumor of the spinal cord. Am J Surg Pathol 21:477–483
Bohinski RJ, Mendel E, Aldape KD, Rhines LD (2004) Intramedullary and extramedullary solitary fibrous tumor of the cervical spine. Case report and review of the literature. J Neurosurg 100:358–363
Burger PC, Scheithauer BW, Vogel FS (2002) Surgical pathology of the nervous system and its coverings, 4th edn. Churchill Livingstone, New York
Cameselle-Teijeiro J, Varela-Duran J, Fonseca E, Villanueva JP, Sobrinh-Simoes M (1994) Solitary fibrous tumor of the thyroid. Am J Clin Pathol 101:535–538
Carneiro SS, Scheithauer BW, Nascimiento AG, Hirose T, Davis DH (1996) Solitary fibrous tumor of the meninges: a lesion distinct from fibrous meningioma. A clinicopathologic and immunohistochemical study. Am J Clin Pathol 106:217–224
Cassarino DS, Auerbach A, Rushing EJ (2003) Widely invasive solitary fibrous tumor of the sphenoid sinus, cavernous sinus and pituitary fossa. Ann Diagn Pathol 7:160–173
Castilla EA, Prayson RA, Stevens GH, Barnett GH (2002) Brain-invasive solitary fibrous tumor of the meninges: report of a case. Int J Surg Pathol 10:217–221
Cummings TJ, Burchette JL, McLendon RE (2001) CD34 and dural fibroblasts: the relationship to solitary fibrous tumor and meningioma. Acta Neuropathologica 102:349–354
Dorfman DM, To K, Dickersin GR, Rosenberg AE, Pilch BZ (1994) Solitary fibrous tumor of the orbit. Am J Surg Pathol 18:281–287
Fletcher CDM (2000) Mesenchymal, non-meningothelial tumors. In: Fletcher CDM (ed) Diagnostic histopathology of tumors, vol 2, 2nd edn. Churchill Livingstone, Philadelphia, pp 1661–1663
Gengler C, Guillou L (2006) Solitary fibrous tumor and hemangiopericytoma: evolution of a concept. Histopathology 48:63–74
Hanau CA, Miettinen M (1995) Solitary fibrous tumor: histological and immunohistochemical spectrum of begnin and malignant variants presenting at different sites. Hum Pathol 26:440–449
Kataoka H, Akiyama Y, Kubo S, Itoh H, Tajima N, Koono M (1999) Solitary fibrous tumor of the spinal nerve rootlet: case report and literature survey. Pathol Int 49:826–830
Klemperer P, Rabin CB (1931) Primary neoplasms of the pleura. Arch Pathol 11:385–412
Kessler A, Lapinsky J, Bernholz L, Sarfaty S, Segal S (1999) Solitary fibrous tumor of the nasal cavity. Otolaryngol Head Neck Surg 121:826–828
Kim KA, Gonzales I, McComb JG, Giannotta SL (2004) Unusual presentations of cerebral solitary fibrous tumors: report of four cases. Neurosurgery 54:1004–1009
Koçak A, çaylI SR, Saraç K, Aydin NE (2004) Intraventricular solitary fibrous tumor: an unusual tumor with radiological, ultrastructural, and immunohistochemical evaluation. Case report. Neurosurgery 54:213–217
Louis DN, Ohgaki H, Wiesler OD, Cavenee WK (2007) WHO classification of tumors of the central nervous system, 4th edn. IARC, Lyon
Metellus P, Bouvier C, Guyotat J, Fuentes S, Jouvet A, Vasiljevic A, Giorgi R, Dufour H, Grisoli F, Figarella-Branger D (2007) Solitary fibrous tumors of the central nervous system: clinicopathological and therapeutic considerations of 18 cases. Neurosurgery 60:715–722
Miyashita K, Hayashi Y, Fujisawa H, Hasegawa M, Yamashita J (2004) Recurrent intracranial solitary fibrous tumor with cerebrospinal fluid dissemination. Case report. J Neurosurg 101:1045–1048
Nakahara K, Yamada M, Shimizu S, Fuji K (2006) Stereotactic radiosurgery as adjuvant treatment for residual solitary fibrous tumor. Case report. J Neurosurg 105:775–776
Pakasa NM, Pasquier B, Chambonnière ML, Morrison AL, Khaddage A, Gentil Perret A, Dumollard JM, Barral FG, Péoc'h M (2005) Atypical presentations of solitary fibrous tumors of the central nervous system: an analysis of unusual clinicopathological and outcomes patterns in three new cases with a review of the literature. Virchows Arch 447:81–86
Perry A, Scheithauer BW, Nascimiento AG (1997) The immunophenotypic spectrum of meningeal hemangiopericytoma: a comparison with fibrous meningioma and solitary fibrous tumor of meninges. Am J Surg Pathol 21:1354–1360
Surendrababu NRS, Chacko G, Daniel RT, Chacko AG (2006) Solitary fibrous tumor of the lateral ventricle: CT appearances and pathologic correlation with follow-up. Am J Neuroradiol 27:2135–2136
Suzuki SO, Fukui M, Nishio S, Iwaki T (2000) Clinicopathological features of solitary fibrous tumor of the meninges: an immunohistichemical reappraisal of cases previously diagnosed to be fibrous meningioma or hemangiopericytoma. Pathol Int 50:808–817
Teranishi K, Yamamoto T, Nakao Y, Osada H, Wada R, Mori K (2007) Recurrent solitary fibrous tumor of the falx cerebri with intraventricular extension. Neurol Med Chir (Tokyo) 47:269–272
Tihan T, Viglione M, Rosenblum MK, Olivi A, Burger PC (2003) Solitary fibrous tumours in the central nervous system. A clinicopathologic review of 18 cases and comparison to meningeal hemangiopericytomas. Arch Pathol Lab Med 127:432–439
Vorster SJ, Prayson RA, Lee JH (2000) Solitary fibrous tumor of the thoracic spine. Case report and review of the literature. J Neurosurg 92:217–220
Young RH, Clement PB, McCaughey WT (1990) Solitary fibrous tumor (‘fibrous mesothelioma’) of the peritoneum. A report of three cases and a review of the literature. Arch Pathol Lab Med 114:493–495
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Comment
This reviewer during his career encountered only one case of histologically verified solitary fibrous tumor (located in the orbit), and read with interest this review of the author's experience. Although this contribution does not contain new information to the existing literature, both the rarity and the atypical presentation of some of the cases illustrated here yet warrant this other publication. I fully agree that the real incidence of such neoplasms may be underestimated.
Domenico d'Avella
Padova, Italy
MRI shows an intensively enhancing and sharply demarcated intracranial tumor:
1. Dural attachment—meningioma/metastasis/malignant glioma ?—but also a hoard of unusual to very rare tumors, including mesenchymal ones (hemangiopericytoma/firbrosarcoma/solitary fibrous tumor or SRT/et al.) and histiocytic ones (Langerhans/Erdham-Chester/Rosai-Dorfman/et al.).
2. Parenchymal or intraventricular—metastasis ?—but also a stock of rare WHO grade I (II) gliomas characterized by intense enhancing and sharp borders other than pilocytic astrocytoma, ganglioglioma, or DNT.
Rare CNS tumors carry an aura of unpredictability. An important part of neurooncology group practice is the recognition of unusual to very rare CNS tumors and the tailoring of optimal therapy and follow-up for their carriers—based on more or less scarce published data. It is axiomatic—never trust the previous diagnosis but have it re-evaluated by your neuropathologist (more than one if in doubt). Neurooncosurgeons are grateful when distinguished colleagues see the trouble of presenting their rare tumors from the last 10 to 20 years, even more so when a critical review of the published cases follows—a practice that should be re-invented in the era of genomics.
The French colleagues do that favor to the readers of Acta Neurochirurgica by presenting their 11 cases of solitary fibrous tumor.
Juha Jaaskelainen
Kuopio, Finland
Rights and permissions
About this article
Cite this article
Vassal, F., Manet, R., Forest, F. et al. Solitary fibrous tumors of the central nervous system: report of five cases with unusual clinicopathological and outcome patterns. Acta Neurochir 153, 377–384 (2011). https://doi.org/10.1007/s00701-010-0866-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00701-010-0866-4