
Abstract
Polyploidization has long been recognized as one of the most important driving forces of plant evolution. Aconitum subgenus Lycoctonum (Ranunculaceae) has a wide distribution range and well-known background of polyploidy, thereby providing a potentially valuable model to explore polyploid origin and evolutionary history. However, the phylogeny of subg. Lycoctonum has not yet been completely resolved. In the current study, 29 species including diploid, tetraploid and hexaploid species were sampled in subg. Lycoctonum. Using four cpDNA regions (ndhF-trnL, psbA-trnH, psbD-trnT and trnT-L) and two nrDNA regions (internal transcribed spacer, ITS, and external transcribed spacer, ETS), phylogenetic relationship was first reconstructed for the polyploid species within subg. Lycoctonum. In combination with nuclear diversification rate estimation, cpDNA haplotype network, ancestral area reconstruction as well as morphological and karyotypic evidence, potential origin and divergence time were further assessed among the polyploid species. Hybridization was inferred for A. angustius and A. finetianum was suggested to be the potential maternal progenitor, due to their close phylogenetic relationship, highly similar morphologies and overlapping distribution range. Local origin was inferred for tetraploids in the Hengduan Mountains (HDM) with eight groups of chromosomes of four homeologous, which diverged approximately 3.00 Ma in the same period of the orogeny of the HDM. The hexaploid A. apetalum was inferred to suffer from geographical isolation due to the formation of the Qinghai–Tibetan Plateau (QTP) and the HDM. Hybridization and heterogeneous habitats in the HDM were suggested to play an important role in the polyploidization in subg. Lycoctonum.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Polyploidization, or whole-genome duplication, has played a critical role in plant speciation and diversification by promoting adaptation to new ecological niches or conferring reproductive isolation (Grant 1971, 1981; Stebbins 1950, 1971; Otto and Whitton 2000; Levin 2002; Soltis et al. 2009; Otto 2007; Paun et al. 2007; Arrigo and Barker 2012). Phylogenetic reconstruction has provided systematists further insights into the assessment of phylogenetic relationships, genome origin and evolution of polyploid species (Soltis and Soltis 1999; Wendel and Doyle 2004; Slotte et al. 2006; Brassac and Blattner 2015). Maternally inherited chloroplast DNA (cpDNA) and biparentally inherited nuclear DNA (nrDNA) sequences have long been treated as a source of phylogenetic data to document origin or evolution of polyploid species (Sharbel and Mitchell-Olds 2001; Abbott and Lowe 2004; Aïnouche et al. 2004; Doyle et al. 2004; Jakobsson et al. 2006; Fortune et al. 2008). Comparison of gene trees derived from different genomes can provide unambiguous evidence of polyploidization and estimate of relative timing without requiring fossil calibration of a tree (Yuan and Olmstead 2008; Yost et al. 2013).
Ranunculaceae represents one of the earliest diverging lineages among the eudicots (APG IV 2016; Wang et al. 2009; Sun et al. 2011). The genus Aconitum L. is a member of the tribe Delphinieae in the Ranunculaceae family. Many species in this genus are proved to be essential components in the formulations of traditional Chinese herbal medicine. Even though some species are highly toxic, they possess variety of medicinal importance (Zhao et al. 2010; Semenov et al. 2016; Wada et al. 2016; Liang et al. 2017). Aconitum currently comprises two subgenera, A. subg. Lycoctonum and A. subg. Aconitum (Jabbour and Renner 2012; Wang et al. 2013). Subgenus Lycoctonum contains approximately 40 species distributed in Eurasia and eastern North America, with approximately 20 species reported in China (Tamura 1995; Gao 2009). Subgenus Lycoctonum differs from subg. Aconitum by its perennial rhizomes, whereas the latter has biennial paired tuberous roots (Tamura and Lauener 1979). Subgenus Lycoctonum has more polyploid members with in-depth investigations of chromosome number, karyotype and geographical distribution (Yang et al. 1994; Yang 1996, 1999, 2001; Gao 2009; Hong et al. 2016). In subg. Lycoctonum, A. angustius (W.T.Wang) W.T.Wang was first revealed to be tetraploid with 32 chromosomes, which are larger than other chromosomes of the tetraploid species in subg. Lycoctonum. The 32 chromosomes can be arranged into eight groups, but four chromosome quartets are heteromorphic with differences in size or morphology (Shang and Lee 1984; Gao et al. 2012; Hong et al. 2016). This tetraploid species was suggested to be widely distributed in southern and central China (Gao 2009; Gao et al. 2012; Hong et al. 2016). An additional four species, including A. brevicalcaratum (Finet and Gagnep.) Diels, A. chrysotrichum W. T. Wang, A. crassiflorum Hand.-Mazz., A. rilongense Kadota, and one variety of A. brevicalcaratum var. parviflorum Chen & Liu, were also determined to be tetraploid (2n = 4x = 32). These five tetraploids are all concentrated in the Hengduan Mountains (HDM) (Yang et al. 1994; Yuan and Yang 2006). The chromosomes of those five tetraploids are similar in size and karyotype constitutions, which are smaller and distinct from that of A. angustius. Eight groups of four homeologous can be fairly well arranged for the chromosomes of the five tetraploids (Hong et al. 2016). The only known hexaploid (2n = 6x = 48) is A. apetalum (Huth) B. Fedtsch with a restricted range in Tianshan Mountains in Xinjiang, northwestern China (Gao 2009). It represents the highest ploidy level currently known in subg. Lycoctonum and is the second polyploid species found to occur outside of the HDM. In summary, a total of six polyploid species and one variety in subg. Lycoctonum have been identified, which are all endemically distributed in China.
Various hypotheses have been proposed in an attempt to interpret polyploid relationships of subg. Lycoctonum based mostly on morphological or cytological evidence. As A. angustius shows heterologous chromosomes as well as intermediate morphological characteristics and overlapping geographical distribution with A. finetianum and A. sinomontanum, hybridization between A. finetianum and A. sinomontanum was suggested for A. angustius by Gao (2009). As for tetraploids endemic to the HDM, they are scarcely distinguishable from one another, particularly A. chrysotrichum, A. brevicalcaratum and A. crassiflorum, showing similar degrees of leaf division, growing habitats and geographical distributions. Yuan and Yang (2006) suggested local origins during the upheaval of the Qinghai-Tibetan Plateau (QTP) for the tetraploids in the HDM. Polyploidization seems to be an important speciation mechanism in the subg. Lycoctonum. However, in the last twenty years, the phylogeny of the subg. Lycoctonum has not yet been accomplished, not to mention the relationships among the polyploid species. Previously, Utelli et al. (2000) suggested early divergence between subg. Lycoctonum and subg. Aconnitum in Europe. High morphological variability within and among populations were detected, most probably due to recent speciation. However, the morphological characters showed no value as systematic characters. Recent phylogenetic reconstruction was conducted within Delphineae, among which nine species from the subg. Lycoctonum were included (Jabbour and Renner 2012). In this study, subg. Lycoctonum was suggested to be monophyletic clade and sister group of Delphinium based on trnL-F and ITS data set (Jabbour and Renner 2012). To the best of our knowledge, no phylogenetic inference has ever been made to infer the relationships between the polyploid species of the subg. Lycoctonum in China.
Uniparentally inherited cpDNA and biparentally inherited nrDNA are frequently combined to make phylogenetic inference, informing relationships among polyploids, origin of hybridization, and the mode of polyploidy in particular groups (Soltis et al. 2004, 2008; Fortune et al. 2008; Kim and Donoghue 2008; Kelly et al. 2013; Dufresne et al. 2014; Brassac and Blattner 2015; Mavrodiev et al. 2015). The nrDNA ITS region is considered concerted evolution of repeated gene families and has been widely analyzed in phylogenetic reconstruction and polyploidization speciation of plants species (Buckler et al. 1997; Álvarez and Wendel 2003; Fortune et al. 2008; Brassac and Blattner 2015). cpDNA is considered to be particularly suitable for investigating processes associated with origin of maternal lineage (Birky 1995; Song et al. 2002). As are motivated to understand phylogenetic relationship and evolutionary history in subg. Lycoctonum, we examined four cpDNA regions with nuclear ITS and ETS sequences from fresh and herbarium material covering a global geographical range, particularly for polyploids in China. Phylogenetic assessment in combination with diversification rate estimation, ancestral area reconstruction and molecular clock dating was used to interpret the phylogenetic relationships and understand the origin of the polyploid species within subg. Lycoctonum. The phylogenetic inference was also determined for the consistence with previous morphological or karyotypic evidence.
Materials and methods
Plant materials
For the present study, we used 48 samples of subg. Lycoctonum representing 26 species and three varieties (Online Resource 1). In particular, we sampled all 16 species and two varieties from China, four species from Europe, three species from Korea, three species (including one variety) from Japan, and the only one species from North America. Three species (A. delavayi, A. tanguticum and A. racemulosum) in A. subg. Aconitum were used as outgroup based on previous studies (Jabbour and Renner 2012; Wang et al. 2013; Luo and Yang 2005). From year 2009 to 2012, we collected individual samples in the field by harvesting young leaves and preserving them in silica gel. All of the voucher specimens of the collected individuals are deposited in the Herbarium of South China Botanical Garden (IBSC). Samples of A. vulparia, A. lycoctonum, A. monticola and A. gigas var. hondoense were obtained from herbarium specimens with the permission of the curators (the first three species from LE and the fourth from PE).
DNA extraction, PCR amplification, and DNA sequencing
Total genomic DNA from approximately 50 mg of fresh dried leaves was extracted using a DNeasy Plant Mini Kit (Qiagen, Hilden, Germany). Six molecular markers, including the cpDNA regions ndhF-trnL, psbA-trnH, psbD-trnT, and trnT-L and the two nrDNA regions, ITS, and ETS, were analyzed. The primer information is listed in Table 1. PCRs were performed in a total volume of 25 μl, containing 14.75 μl sterile purified water, 5.0 μl 5 × PrimeSTAR® Buffer (Mg2+ plus), 2.0 μl dNTP Mixture (2.5 mM each nucleotide), 0.5 μl of each primer (10 μM), 0.25 μl PrimeSTAR® HS DNA Polymerase (2.5 U/μl), and 2.0 μl DNA template (ca. 50 ng). The PCR protocol included an initial denaturation (3 min at 95 °C) followed by 32 cycles of 30 s denaturation (94 °C), 40 s annealing (52–56 °C), and 1 min extension at 72 °C, and concluded with a final extension of 8 min at 72 °C. All PCR products were purified using a TIANquick Midi Purification Kit (TIANGEN, Beijing, China) and sequenced using the corresponding PCR primers. We obtained sequences from an ABI PRISM 3100 Genetic Analyzer. We deposited all sequences in GenBank (accession numbers shown in Online Resource 1).
Sequence alignment and phylogenetic analyses
All sequence alignments were conducted using MAFFT v.6.717 (Katoh and Frith 2012) components from SATéwin v.2.2.7-2013 Feb15 (Liu et al. 2012). Alignment improvements were conducted manually by eye using BioEdit v.7.0.9.0 (Hall 1999). The complete combined alignment has been deposited in TreeBASE (http://treebase.org/treebase-web/home.html) with accession number S20706. Gaps were treated as missing data in the alignments and in downstream analyses. To determine the potential of combining the cpDNA and nrDNA sequence datasets for the phylogenetic analyses, we used Incongruence Length Difference (ILD) tests (Farris et al. 1995) implemented in PAUP* v.4.0b10 (Swofford 2002). The ILD tests were conducted using 1000 replicates and random addition (10 additional sequences), maintaining a maximum of 100 trees in each repetition. We considered P < 0.001 as a more realistic criterion to reject combinability (Cunningham 1997a). In particular, congruence was tested among the molecular markers and between combined cpDNA and nrDNA data sets, which were used separately and in combination for further phylogenetic analyses.
We performed a Bayesian Inference (BI) of phylogenies using MrBayes v.3.2 (Huelsenbeck and Ronquist 2001; Ronquist and Huelsenbeck 2003). Modeltest v.3.7 (Posada and Crandall 1998) was used to select from 24 models of nucleotide substitution to determine the best model for each sequence partition. Four simultaneous independent runs were performed with four chains each (one cold and three heated) and initiated with random trees. We unlinked the data partitions during the analyses so each partition had independently estimated parameters, and we saved one tree every 1000 generations. All BI analyses reached an average standard deviation of split frequencies ≤0.01 after 20 million generations, and we excluded 25% of the generated trees as burn-in. To confirm the standard deviations and burn-in values, we assessed convergence and stationarity, respectively, by examining the split frequencies diagnostic and by comparing the mean and variance of the log likelihoods in the “.stat” output file from MrBayes. We used the remaining trees from the four runs to estimate the 50% majority-rule consensus tree and the Bayesian posterior probabilities. Maximum Parsimony (MP) was performed using PAUP* v.4.0b10 (Swofford 2002). All characters were treated as equally weighted. Heuristic searches included 1000 replicates with random sequence additions, tree bisection-reconnection (TBR) branch swapping, Multrees on, and maxtrees set to 10 for each step. We assessed clades supported by the MP analyses with bootstrap values (Felsenstein 1985). Bootstrap values were calculated in PAUP* with 1000 bootstrap replicates, each with 10 random sequence addition replicates holding maximally one tree per replicate, TBR branch swapping, and Multrees on. Maximum likelihood (ML) analyses used the identical partition scheme and were conducted using RAxML (Stamatakis 2006) and the RAxML graphical interface (raxmlGUI v.1.3 (Silvestro and Michalak 2012)). RAxML was conducted using Python v.2.7.6 (http://www.python.org/ftp/python/2.7.6/python-2.7.6.msi) with 1000 rapid bootstrap replicates. For the ML analyses, the general time-reversible (GTR) model was used with a gamma model of the rate of heterogeneity. Additionally, to visualize the conflicts among the gene trees, the FilteredSuperNetwork implemented in SplitsTree4 v4.13.1 (Huson and Bryant 2006) was used to generate a consensus tree of the separate MP trees (Four cpDNA and two nrDNA regions). The input trees were edited as multiple individuals in a monophyletic clade collapsed to one terminal. The default settings were used to construct the network.
cpDNA haplotype network analysis
To detect representative groups of polyploids, a genealogical degree of relatedness among the cpDNA haplotypes of subg. Lycoctonum was estimated using TCS v.3.11 (Clement et al. 2000) based on a maximum parsimony approach, performing best for non-recombinant sequences (Woolley et al. 2008). In this analysis, indels were treated as single mutation events regardless of their length. Different types of insertions occurring at one site were coded as one of four nucleotides. In both the parsimonious and network analyses, length variations in mononucleotide repeats (poly A or T) were excluded because of their tendency for homoplasy.
Reconstruction of the ancestral area
For ancestral area reconstruction, the distribution of subg. Lycoctonum was divided into eight areas: northwestern China (A), Qinghai–Tibetan Plateau (QTP; B), Hengduan Mountains (HDM; C), central China (D), eastern Asia (E), central Asia (F), Europe (G) and North America (H) (Fig. 1). Korea and Japan were included in Eastern Asia, as they were connected via the Eastern Sea Land Bridge during the Pleistocene glaciations (Harrison et al. 2001; Sun et al. 1999; Qiu et al. 2011). Statistical dispersal-vicariance analysis (S-DIVA) and Bayesian binary MCMC (BBM) programs implemented in RASP v.3.2 (Yu et al. 2015) were used to reconstruct the possible ancestral areas of subg. Lycoctonum on the combined consensus ML tree generated by RAxML (Stamatakis 2006), which was the dataset used for the divergence time analysis. In this method, the frequencies of an ancestral area at a node in the ancestral reconstructions are averaged over all trees. To account for uncertainties in the phylogeny, we used 3000 trees from the MCMC output and ran a BBM analysis on the condensed tree. The ten MCMC chains were run simultaneously for 500,000 cycles, and the state was sampled every 100 generations. A fixed Jukes-Cantor model with equal among-site rate variation was used for the BBM analysis with root distribution set to “outgroup.” The maximum number of areas for this analysis was six. The dispersal and vicariance events were inferred using a dispersal-vicariance analysis (Ronquist 1997) implemented in RASP v.3.2 (Yu et al. 2015).

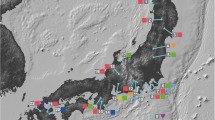
Map of geographical distribution and sample localities of subg. Lycoctonum species both inside and outside China. Circles with numbers 1–29 represent the species corresponding with Online Resource 1. Diploid, tetraploid and hexaploid species are each indicated by circles filled in three different colors of gray, orange and blue. Circles outlined with eight different colors represent eight different geographical distribution regions of subg. Lycoctonum, corresponding to S-DIVA analysis. Gray area with black outline shows the geographical distribution range of Aconitum sinomontanum, orange area with orange outline for A. angustius, and gray areas with orange outline for A. finetianum. Gray straight lines and dotted lines with arrows indicate the vicariance and dispersal events, respectively, by S-DIVA analysis
Molecular clock estimation and diversification rate analyses
We analyzed the divergence time in BEAST v.1.8.0 (Drummond et al. 2006, 2012) for a combined dataset of 51 subg. Lycoctonum samples and the outgroup with 2915 aligned nucleotides. The divergence time analyses were conducted using a GTR+G model with four rate categories, a Yule tree prior, and relaxed clocks with uncorrelated and a log-normally distributed rate variation across the branches. We ran the MCMC simulations in BEAST for 20 million generations with parameters sampled every 2000th step. At the conclusion of the analysis, we applied a burn-in of 10%. We combined the results from individual runs and used Tracer v.1.6 (Rambaut et al. 2013) to check that the effective sample sizes for all relevant parameters were well above 200 and stationary. The final trees were edited using FigTree v.1.4.0 (http://tree.bio.ed.as.uk/software/figtree/).
Secondary calibration points are divergence time estimates that have been derived from one molecular dataset on the basis of a primary external calibration point and are used again independently of the original external calibration point on a second dataset. As Ranunculaceae fossils were not available to use as calibration points (Pigg and DeVore 2005; Jabbour and Renner 2011), Jabbour and Renner (2012) used the cross-validated calibration approaches to analyze the diversification of Ranunculaceae. They further used those result as secondary calibrations for the phylogenetic analyses of Delphinieae, which should offer more precise divergence time to serve as calibration points. In the same way, we used the result of Jabbour and Renner (2012) as calibration points, in which Aconitum was shown to evolve 12.70 Ma and the crown node of subg. Aconitum began diversifying 7.80 Ma. Therefore, we used those two ages as calibration points with normal prior distributions and standard deviations of 2.20 Ma, which covered the 95% HPD range. We attended to the secondary calibrations’ limitations, which tend to result in younger ages compared to those obtained with fossils (Sauquet et al. 2012).
For the diversification rate analyses, we excluded three species of the outgroup, five species outside of China or eastern Asia and two varieties, to assure a broad taxonomic sampling in China and eastern Asia (Deng et al. 2015). One random sample from each species was used for the following analyses. The temporal dynamics of diversification in subg. Lycoctonum was measured using LTT plots in the APE package of R (Paradis et al. 2004). To compare the diversification rates, the net diversification rates were estimated for different lineages and regions. Plots were produced based on the branching time information obtained from the BEAST analysis. The GEIGER package in R (Harmon et al. 2008) was used to calculate the net diversification rates (γ) of subg. Lycoctonum based on the equation (7) of Magallón and Sanderson (2001) (with no extinction ε = 0 and high extinction rate ε = 0.90). However, the BAMM v.2.2.0 program was also applied to explore the diversification rate heterogeneity among lineages in different geological periods (Rabosky 2014). The analysis was run with 10 million MCMC generations, which were sampled every 2000 generations. The convergence was tested based on the MCMC output using the CODA package in R (Plummer et al. 2006) with a burn-in of 10% and effective sample sizes exceeding 1000 for all estimated parameters. The BAMM output was used to calculate diversification rates with BAMMtools v.2.0.2 in R (Rabosky et al. 2014). The 95% credible shift configurations were filtered using a Bayes factor of 5, and the mean phylorate for net diversification, and marginal probabilities were plotted with the getEventData and plot.bammdata functions.
Results
Sequence characterization, ILD test and phylogenetic reconstruction
Forty-eight samples representing 26 species and three varieties of subg. Lycoctonum, and three samples from outgroup species were analyzed for both the cpDNA and nrDNA data sets. For the cpDNA dataset, samples collected from herbarium specimens were able to be amplified successfully. In Table 2, the aligned length of four cpDNA regions varied from 302 to 917 bp. The ambiguously aligned characters of the cpDNA dataset were excluded from all analyses. Therefore, 2071 aligned nucleotides were generated. A total of 174 bp (8.40%) were variable and 71 bp (3.43%) were parsimony-informative loci in the cpDNA dataset. The lengths of the ITS and ETS were 518 and 326 bp, respectively. The combined nrDNA sequences consisted of 844 aligned nucleotides, of which a total of 254 bp (30.09%) were variable and 162 bp (19.19%) were parsimony informative. The ILD test showed that the sequences within the cpDNA and nrDNA datasets, independently, were highly congruent (P > 0.01 for cpDNA; P = 0.42 for nrDNA). While the ILD test detected that sequences between the cpDNA and nrDNA datasets are incongruent (P = 0.001). The combination of the two incongruent matrixes produced largely congruent topologies with the filtered supernetwork, generated by the consensus tree of the separate MP trees (Fig. 2; Online Resource 2–4). Therefore, we used the combined two matrixes to improve phylogenetic accuracy (Cunningham 1997b; Yi et al. 2014). For the combined matrix of the cpDNA and nrDNA datasets, 2915 bp aligned nucleotides were detected containing 196 (7.30%) parsimony-informative sites. With the combined sequences, the MP analysis recovered 1000 most parsimonious trees of 625 steps with consistency index (CI) and retention index (RI) values of 0.77 and 0.89, respectively. The tree topologies obtained from the MP, BI and ML analyses, based on the cpDNA and nrDNA datasets, are largely congruent (Online Resource 2–3). Because the filtered supernetwork also showed largely congruent topologies with a combined ML tree in subg. Lycoctonum, the MP, BI, and ML analyses, based on combined datasets and the filtered supernetwork, will be discussed together.

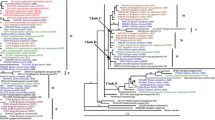
The best-scoring maximum likelihood (ML) tree inferred from the combined cpDNA and nrDNA data sets with the GTRGAMMA model implemented in RAxML. The numbers near the branches are Bayesian posterior probabilities and bootstrap percentages of maximum parsimony and maximum likelihood trees, respectively. Dashed lines indicate nodes with less than 50% support. Species with multiple individuals are represented by the abbreviations of voucher numbers. Two major clades were identified (clade 1 and clade 2), each with three subclades. The tetraploid species, hexaploid species and the potential diploid ancestor are all highlighted with shades in three different colors of orange, blue and gray. Species distributed in the Hengduan Mountains (HDM), Korea and Japan are all indicated
Two diploid species, A. novoluridum and A. fletcheranum, were positioned outside of the major clades at the base branches in the consensus tree compiled from ML with high support values (100/95/99 BI/MP/ML and 100/100/100 BI/MP/ML, respectively). Two major clades (clade 1 and clade 2) were detected, each of which was divided into three subclades (Fig. 2). The first clade (clade 1), with three subclades 1–1, 1–2 and 1–3, was composed of tetraploid A. angustius and all diploid species except A. scaposum. Aconitum angustius, A. finetianum and A. alboviolaceum in eastern and northeastern China, three Korean and two Japanese species together formed subclade 1–1, showing monophyly with strong support (100/100/99 BI/MP/ML). However, the inner support for the deeper nodes was weaker except for the three Korean species (99/68/91 BI/MP/ML). In subclade 1–1, A. angustius was more frequently present with A. finetianum (Fig. 2). Subclade 1–2 was composed of diploids, three from Europe and six from China with strong support (100/99/97 BI/MP/ML). Aconitum sinomontanum is placed in subclade 1–2. Subclade 1–3 consisted of diploid species each from Japan, North America and central Asia and three diploid species from China with strong support (99/82/89 BI/MP/ML). The second clade (clade 2) had three subclades, 2–1, 2–2 and 2–3, and was composed of all tetraploids in the HDM, the hexaploid A. apetalum and widespread diploid species A. scaposum. Moderate support values (65/69/79 BI/MP/ML) were detected for clade 2, but strong support values were detected for the subclades. Subclade 2–1 was formed by all tetraploids in the HDM and showed the high support values of 100/95/98 BI/MP/ML. Aconitum apetalum and A. scaposum were clustered into separate subclades 2–2 and 2–3, respectively, with full support in all analyses.
Three species, A. gigas var. hondoense, A. scaposum and A. apetalum, showed incongruent positions based on partial datasets (Online Resource 2–3). The position of A. gigas var. hondoense changed from subclade 1–1 based on cpDNA data to subclade 1–3 based on nrDNA data. However, no support values were detected in either partial dataset for the placement of A. gigas var. hondoense (Online Resource 2–3). The positions of A. scaposum and A. apetalum, which were clustered with tetraploids in the HDM by the cpDNA data (80/73/88 BI/MP/ML), had extremely low support (51/-/- BI/MP/ML) as a sister to all other species in clade 1 based on the nrDNA data (Online Resource 2–3).
cpDNA haplotype network
Forty haplotypes (H1–H40) were identified among the four cpDNA regions for 26 species and three varieties, of which 37 were species-specific (Online Resource 1; Fig. 3). Haplotype H2 was present in both A. angustius and A. finetianum, haplotype H5 shared between A. barbatum, A. barbatum var. puberulum, A. shennongjiaense and A. sinomontanum, and H39 occurring in two Korean species, A. longecassidatum and A. quelpaertense. Two cpDNA haplotype groups, Group 1 and Group 2, consisting of polyploidy species, were identified in the TCS network, which largely corresponded with subclade 1–1 and clade 2, respectively (Fig. 2). In haplotype Group 1, A. angustius (H1, H2, H28 and H31) was positioned at the end step and was closely connected with A. finetianum (H2, H10 and H11) and A. alboviolaceum (H29 and H30). Two Korean haplotypes (H39 and H40) were derived from haplotype H30 of A. alboviolaceum. The Japanese species were represented by three haplotypes H13, H18 and H38, of which H38 was six steps from A. alboviolaceum (H29). In haplotype Group 2, all tetraploids (H6–H9, H20, and H32–H37) in the HDM were connected with A. scaposum (H21, H22 and H23) and A. apetalum (H3 and H4). Ancestry missing haplotypes were detected for species in haplotype Group 2.

The 95% plausible cpDNA haplotypes network inferred by TCS. Forty cpDNA haplotypes are shown. The size of each circle corresponds to the frequency of each haplotype. Dots with black outlines indicate unsampled or extinct haplotypes. The diploid, tetraploid and hexaploid species are all highlighted with three different colors of gray, orange and blue. Two cpDNA haplotype groups (1–2) consisting polyploid species were identified. Species distributed in Hengduan Mountains (HDM), Korea and Japan are all indicated
Ancestral area reconstruction
S-DIVA indicated an obvious biogeographical history for polyploidy species in subg. Lycoctonum. Altogether, nine vicariance and seven dispersal events with Bayesian probabilities (P values) ≥ 0.70 were postulated (Figs. 1, 4). In subclade 1–1, three vicariance events (V1, V2 and V3; P = 1.00) were identified between central China (D) and eastern Asia (E). V1 (P = 1.00) was detected at the node where the Korean species diverged from A. angustius and A. finetianum. Two dispersal events (D1 and D2; P = 1.00) were detected showing a route from eastern Asia (E) to central China (D) and back to eastern Asia (E). Eastern Asia (E) was considered to be the ancestral area (AA1) for subclade 1–1. In clade 2, two vicariance events (V4 and V5; P = 1.00) were identified, of which V5 (P = 1.00) was between the HDM (C) and northwestern China (A). Two dispersal events (D3 and D4; P = 1.00) were detected showing a routes from the HDM (C) to northwestern China (A) and central China (D) with the HDM (C) being the center of origin (AA2).

Ancestral area reconstruction of subg. Lycoctonum species as inferred by S-DIVA. Color legends indicated eight geographical distribution regions (A–H) and other ancestral area. The eight geographical distribution regions (A–H) assigned for each sample, the numbers (1–29) representing the species and the ploidy levels of each sample are all given at the bottom. Vicariance events (V1–V9) and dispersal events (D1–D7), with Bayesian probabilities (P values) ≥0.70, are shown for nodes. Ancestral areas (AA1 in region E and AA2 in region C) are indicated for subclade 1–1 and subclade 2–1 consisting polyploidy species, respectively
Divergence time estimation and diversification rate analyses
According to Fig. 5, the calculated divergence time of subg. Lycoctonum was estimated as 10.63 (95% HPD = 6.70–15.00) Ma during the middle Miocene. The divergence time between clade 1 and clade 2 was inferred as 5.72 (95% HPD = 3.23–8.56) Ma in the late Miocene. Subclade 1–1 was estimated to diverge approximately 2.24 (95% HPD = 1.03–3.73) Ma in late Pliocene. The ancestor of clade 2 was estimated to arise approximately 5.13 (95% HPD = 2.74–7.74) Ma in the late Miocene. Tetraploids in the HDM, forming subclade 2–1, were estimated to have diversified approximately 3.00 (95% HPD = 1.44–4.90) Ma and diverged from A. apetalum approximately 4.13 (95% HPD = 2.19–6.48) Ma in mid-Pliocene. Compared to the results of Jabbour and Renner (2012), the estimated divergence ages were younger because of the limitations of the secondary calibrations (Sauquet et al. 2012).

Chronogram for the combined dataset obtained under a Bayesian relaxed clock with log-normally distributed rates. The 95% highest posterior density (HPD) for major lineages with a posterior probability >0.95 are indicated at the nodes. Time scale is shown on the bottom. The numbers corresponded to the code numbers in Table 1. Chromosome numbers are indicated following the species names. Two major clades were identified (clade 1 and clade 2), each with three subclades. The tetraploid species, hexaploid species and the potential diploid ancestor are all highlighted with shades in three different colors of orange, blue and gray. Species distributed in the Hengduan Mountains (HDM), Korea and Japan are all indicated
In particular, the LTT analysis suggested an increasement in the diversification rate of subg. Lycoctonum at approximately 6.35 Ma in the late Miocene (Fig. 6). The net diversification rates of subg. Lycoctonum were 0.24 sp/Myr (ε = 0) and 0.11 sp/Myr (ε = 0.90) (Table 3). The diversification rates of clade 1–1 were 0.8402 sp/Myr (ε = 0) and 0.34 sp/Myr (ε = 0.90) (Table 3). The diversification rates of clade 2–1 were 0.26 sp/Myr (ε = 0) and 0.07 sp/Myr (ε = 0.90), which were much lower than other subclades (Table 3). The BAMM analysis also indicated a high heterogeneity of diversification rates among different lineages during different geological periods. Subclade 2–1 showed a comparably lower diversification rate in the early Pliocene (Fig. 7). The species diversification rate in the HDM was obviously lower than other regions (Table 3; Fig. 7).

Log lineage-through-time plot for the subg. Lycoctonum

A phylorate plots showing the heterogeneity of diversification rates in subg. Lycoctonum estimated from BAMM
Discussion
The phylogenetic relationship and diversification rate of subgenus Lycoctonum
The ILD test detected incongruence between the sequences of the cpDNA and nrDNA datasets. Phylogenetic incongruence between genes is widespread and expected because unlinked genes have different evolutionary histories (Huelsenbeck et al. 1996; Mansion et al. 2005; Lihova et al. 2006). According to Cunningham (1997a), P < 0.001 is a more realistic criterion to reject combinability. However, studies have previously shown that even when the ILD test indicates incongruence between datasets, the combined analysis can provide better resolved trees (Camacho-García et al. 2014; Oskars et al. 2015).
Major phylogenetic relationships within subg. Lycoctonum were resolved and generally well supported in the current study. Remarkably, relationships among the subclades were also strongly supported. Different methods (MP, BI, ML and a filtered supernetwork) and datasets (cpDNA and nrDNA) generated largely consistent phylogenetic tree. Aconitum fletcheranum and A. novoluridum, which are distributed in Qinghai-Tibetan Plateau (QTP), were revealed at the base branches and showed distant relationship with other species in subg. Lycoctonum. Aconitum fletcheranum is the only species displaying terminal inflorescence (Tamura 1995), and it represents a different base chromosome numbers in subg. Lycoctonum (Langlet 1927; Gregory 1941; Kurita 1957; Tamura 1995; Yuan and Yang 2006; Hong et al. 2016). The karyotype of A. novoluridum mainly consists of m (median centromeric) and sm (submedian centromeric) chromosomes, remarkable different from other diploid species mainly consisting of st (subterminal centromeric) chromosomes (Yuan and Yang 2006). Additionally, previous karyotypic analysis and taxonomic studies suggested the two species as two independent sections (Tamura 1995; Gao 2009). We inferred that the two phylogenetically distant species have suffered from a long-term geographical isolation, contributing to their unique chromosome characteristics and distant phylogenetic positions. The widespread A. scaposum was also supported to be independent Ser. Scaposa W. T. Wang (Gao 2009), which is consistent with the phylogenetic result. The position of the Japanese species A. gigas var. hondoense remains uncertain because it showed alternative and inconsistent clustering without support values based on both datasets (Online Resource 2–3).
Aconitum finetianum, A. aloviolaceum and other Korean and Japanese species formed monophyly with A. angustius (subclade 1–1). The diploid species in subclade 1–1 displayed extensive morphological variation with no relation to species or geographical distribution, often leading to misidentification among them. For example, A. finetianum and A. longecassidatum, with minor differences in sepal hairs, were previously treated as conspecific by Tamura and Lauener (1979). Aconitum pterocaule was previously thought to be synonymous with A. finetianum and A. longecassidatum (Tamura and Lauener 1979), which were present in Eastern Asia, including Korea Peninsula, Japan Island as well as northeastern and eastern China (Fig. 1). These areas were geographically connected via the Eastern Sea Land Bridge during the Pleistocene glaciations (Sun et al. 1999; Harrison et al. 2001; Qiu et al. 2011). Meanwhile, this geographical region exhibits a little bit flat topography and has experienced multi times of climatic oscillation. Therefore, gene introgression could be facilitated and resulted in extensive morphological similarities and close phylogenetic relationships among those diploid species. Aconitum angustius shows intermediate morphological characteristics between A. finetianum and A. sinomontanum. It is reasonable to understand that A. angustius was closely related with A. finetianum on the phylogenetic tree. However, it is not expected that A. angustius demonstrated distant positions from A. sinomontanum. This is most probably due to the limitation of the uniparentally inherited cpDNA regions, which might not be able to reveal the paternal lineages among species. Aconitum sinomontanum was arranged in Ser. Lycoctonia Tamura & Lauener with A. shennongjiaense, A. monticola, and A. umbrosum based on taxonomical analysis (Gao 2009). The taxonomical result was further supported by the current phylogenetic tree.
All the tetraploid species in the HDM formed one monophyly, supporting them to be classified into Ser. Crassiflora Tamura & Lauener (Gao 2009). Only minor divergences could be identified for the morphological characteristics, living habitats and geographical distribution among the tetraploid species in the HDM. This might suggest that all the tetraploid species in the HDM originated from the same ancestor with a rapid radiation of species diversity. The HDM is dominated by complicated topography with deep and parallel valleys allowing extreme elevation changes within relatively short distances, thus producing local habitat uniqueness and plant species richness. It was also suggested to be a long-term refugium for a few species with ancient origins. Various examples for rapid radiation and glacial refugium of the HDM could be found in Magacodon styrophorus (Ge et al. 2005), Ligularia tongolensis (Wang et al. 2011) and some cypress species (Xu et al. 2010). The hexaploid A. apetalum, showing the highest ploidy level in subg. Lycoctonum, is known to be geographically isolated and distributed in central Asia. Taxonomically, Gao (2009) set it as an independent Ser. Micrantha Steinb. ex Tamura & Lauener due to the different upper sepal characteristics from other tetraploid species in the HDM. Consistently, it was revealed to be a monophyly based on the current phylogenetic tree. In chromosome size and particularly chromosome morphology, A. apetalum showed somewhat similar characteristics to the tetraploid species in the HDM, but markedly different from A. angustius (Hong et al. 2016). Therefore, it was supported as the sister group of the tetraploid species in the HDM.
The overall diversification rates were 0.24 sp/Myr (ε = 0) and 0.11 sp/Myr (ε = 0.90) for subg. Lycoctonum, which were different from those (0.61 sp/Myr (ε = 0) and 0.33 sp/Myr (ε = 0.90)) detected by Jabbour and Renner (2012). The difference in diversification rates in the two studies resulted from different species sampling, as relatively fewer ancient species in subg. Lycoctonum were sampled in Jabbour and Renner (2012) than in the current study. Polyploidy drives speciation and diversification as is evident in many angiosperm families with origins of many major clades being preceded by whole-genome duplications (Stebbins 1971; Otto and Whitton 2000; Levin 2002; Otto 2007; Paun et al. 2007; Soltis et al. 2009; Arrigo and Barker 2012; Estep et al. 2014). However, no obvious correlation between polyploidization and the subsequent increase in net diversification rate were suggested based on theoretical arguments and empirical studies most recently (Wood et al. 2009; Mayrose et al. 2011; Estep et al. 2014). In the current study, the lineages containing A. angustius (subclade 1–1) demonstrated a lower diversification rate than subclade 1–2. In addition, the lineage containing tetraploids in the HDM (subclade 2–1) showed a much lower diversification rate than the other lineages (Table 3; Fig. 7). Studies have shown that a high diversification rate does not always occur in areas with noted biodiversity (Escudero et al. 2012; Christin et al. 2014; Schilling et al. 2015), such is the case in the current study. The unanticipated low diversification rate occurred in the HDM, where there is high species diversity. This might have been produced by the identically local origin in the HDM.
Phylogenetic relationships among the polyploid species
Interspecific hybridization and subsequent genome doubling were suggested to occur frequently as one of the mechanisms of polyploid speciation (Thompson and Lumaret 1992; Soltis and Soltis 1999; Ramsey and Schemske 1998, 2002). Usually hybridization is resulted from different diploid species and thus leading to polyploid speciation. Cytological investigations have shown that A. angustius possesses 32 chromosomes mainly with st types, which are larger and cannot be arranged into eight groups of four homologs as other tetraploid species in the HDM (Gao 2009; Gao et al. 2012; Hong et al. 2016). Thus, tetraploidy in A. angustius could not have arisen from a single progenitor and most likely resulted from hybridization. Morphologically, A. angustius displays intermediate morphologies between A. finetianum and A. sinomontanum. It shows morphological similarities in having leaves 3-parted, pedicels retrosely pubescent and the upper sepal cylindric with A. finetianum, often making them confused with each other (Gao et al. 2012). Meanwhile, A. angustius often displays overlapping geographical ranges with A. finetianum and A. sinomontanum. For example, it co-occurs with A. finetianum on several mountains in southeast China (Lushan Mountains, Hengshan Mountains). Especially, on Lushan Mountains, they even grow side by side within a community (Yuan and Yang 2006). Its northern margin of the geographical distribution range overlapped with A. sinomontanum in western Hubei and northeastern Chongqing (Fig. 1). However, the current study only indicated close phylogenetic relationship (Fig. 2) and common shared cpDNA haplotype H2 between A. finetianum and A. angustius (Table 1; Fig. 3). In combination with morphological, cytological and phylogenetic evidence, we therefore suggest A. finetianum as the potential maternal progenitor of A. angustius. Though A. sinomontanum was previously suggested to be another progenitor of A. angustius, no molecular evidence was available in the current study. This is most probably due to the limitation of the uniparentally inherited cpDNA regions and concerted evolution of nrDNA ITS region. The relationships of A. angustius and A. sinomontanum need further determination.
Heterogeneous habitats were suggested as geographical regions for rapid diversification and long-term preservation of plant species (Tuomisto et al. 2003; Kreft et al. 2006; Antonelli and Sanmartín 2011; Verboom et al. 2015). Tetraploids in the HDM displayed similar morphological characteristics and karyotypes (Yuan and Yang 2006; Hong et al. 2016) and formed monophyly in the phylogenetic tree. Those species can only be distinguished by minor differences of sepals and spurs. Their geographical distribution range and growing habitats were hard to be differentiated. Karyotypic constitutions showed mainly sm or m types of those species, which are smaller than that of A. angustius and can be arranged fairly well into eight groups of four homologs. All above evidence supported the same origin of the five tetraploid species in the HDM. The geographical center of the local origin was detected in the HDM (C) (AA2) (Fig. 4), where the tetraploids were estimated to originate approximately 3.00 (95% HPD = 1.44–4.90) Ma in the middle Pliocene (Fig. 5). This period was coincident with the orogeny of the HDM (approximately 3.4 Ma) (Li et al. 1979, 1996; Cui et al. 1996; Shi et al. 1998; Wang et al. 2010; Liu et al. 2013). We therefore suggest local origin in the HDM of the five tetraploids. Perennial growth habit is considered to be one of the factors promoting polyploidy (Grant 1971, 1981). In the HDM, the uplift of the QTP and climate change could have led to high levels of vegetative propagation (e.g., unreduced gametes), thus resulting in local species diversification. Meanwhile, the HDM is dominated by heterogeneous habitats, showing deep and parallel valleys with extreme elevation changes within relatively short distances. It is considered as an important geographical region for rapid radiation of plant species (Li and Li 1993; Wu 1988; Myers et al. 2000), which facilitated species diversification and long-term preservation. However, the progenitors of tetraploids in the HDM were unable to be detected due to the missing cpDNA haplotypes (Fig. 3).
Aconitum apetalum is the only hexaploid species in subg. Lycoctonum, which is distributed in the Tianshan Mountains, Xinjiang, northwestern China and Kazakhstan and Tajikistan in central Asia. It demonstrated similar karyotypic constitutions and close phylogenetic relationship with the tetraploid species in the HDM. In the present study, A. apetalum was estimated to originate approximately 4.13 (95% HPD = 2.19–6.48) Ma, which was earlier than the orogeny of the HDM (approximately 3.40 Ma) and the fourth period of the uplift of the QTP (approximately 1.60–3.50 Ma) (Liu et al. 2013; Harrison et al. 1992; Chen 1992; Spicer et al. 2003; Wen et al. 2014). A vicariance event (V5; P = 1.00) was also detected between the HDM (C) and northwestern China (A) according to the S-DIVA inference (Fig. 4). Therefore, it is reasonable to infer that A. apetalum has suffered strongly from long-term geographical isolation or forced migration northwards resulting from the formation of geographical barriers by the HDM and QTP.
Conclusions
Subgenus Lycoctonum has a widespread range and a well-known background of polyploidy. It is a diverse subgenus in one of the earliest diverging lineages of Ranunculaceae, composed of diploid, tetraploid as well as hexaploid species. However, phylogenetic assessment has never been well accomplished in particularly for the polyploid species. In the current study, we examined cpDNA regions and nrDNA sequences from subg. Lycoctonum species. Phylogenetic inference was conducted informing the relationship between the polyploid species. Major phylogenetic relationships within subg. Lycoctonum were resolved with generally high support values and consistence with previous taxonomic studies. No evidence of polyploidization leading to an increased diversification rate was observed. Ancestral area was reconstructed, and divergence time was estimated in particular for the polyploid lineages. Hybridization was suggested for A. angustius and A. finetianum was inferred to be the potential maternal progenitor. Local origin was suggested for tetraploids in the HDM. Heterogeneous habitats were considered to be important factors promoting rapid speciation and long-term preservation in the HDM. Evidently, A. apetalum has suffered from long-term geographical isolation by the QTP and HDM. Therefore, hybridization and heterogeneous habitats have played an important role in the polyploidization of subg. Lycoctonum. However, more molecular evidence is in need for further interpretation of the phylogenetic relationship and polyploids evolutionary mechanism of subg. Lycoctonum.
References
Abbott RJ, Lowe AJ (2004) Origins, establishment and evolution of new polyploid species: Senecio cambrensis and S. eboracensis in the British Isles. Biol J Linn Soc 82:467–474. doi:10.1111/j.1095-8312.2004.00333.x
Aïnouche ML, Baumel A, Salmon A (2004) Spartina anglica C.E. Hubbard: a natural model system for analyzing early evolution changes that affect allopolyploid genomes. Biol J Linn Soc 82:475–484. doi:10.1111/j.1095-8312.2004.00334.x
Álvarez I, Wendel JF (2003) Ribosomal ITS sequences and plant phylogenetic inference. Molec Phylogen Evol 29:417–434. doi:10.1016/S1055-7903(03)00208-2
Antonelli A, Sanmartín I (2011) Why are there so many plant species in the Neotropics? Taxon 60:403–414
APG IV (2016) An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot J Linn Soc 181:1–20
Arrigo N, Barker MS (2012) Rarely successful polyploids and their legacy in plant genomes. Curr Opin Pl Biol 15:140–146. doi:10.1016/j.pbi.2012.03.010
Baldwin BG, Markos S (1998) Phylogenetic Utility of the External Transcribed Spacer (ETS) of 18S–26S rDNA: Congruence of ETS and ITS Trees of Calycadenia (Compositae). Molec Phylogen Evol 10:449–463. doi:10.1006/mpev.1998.0545
Birky CW (1995) Uniparental inheritance of mitochondrial and chloroplast genes mechanisms and evolution. Proc Natl Acad Sci USA 92:11331–11338. doi:10.1073/pnas.92.25.11331
Brassac J, Blattner FR (2015) Species-level phylogeny and polyploid relationships in Hordeum (Poaceae) inferred by next-generation sequencing and in silico cloning of multiple nuclear loci. Syst Biol 64:792–808. doi:10.1093/sysbio/syv035
Buckler ES, Ippolito A, Holtsford TP (1997) The evolution of ribosomal DNA: divergent paralogues and phylogenetic implications. Genetics 145:821–832
Camacho-García YE, Ornelas-Gatdula E, Gosliner TM, Valdés Á (2014) Phylogeny of the family Aglajidae (Pilsbry, 1895) (Heterobranchia: Cephalaspidea) inferred from mtDNA and nDNA. Molec Phylogen Evol 71:113–126. doi:10.1016/j.ympev.2013.11.010
Chen FB (1992) H-D event: an important tectonic event of the late Cenozoic in Eastern Asia. Mountain Res 10:195–202
Christin PA, Spriggs E, Osborne CP, Strömberg CAE, Salamin N, Edwards EJ (2014) Molecular dating, evolutionary rates, and the age of the grasses. Syst Biol 63:153–165. doi:10.1093/sysbio/syt072
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Molec Ecol 9:1657–1660. doi:10.1046/j.1365-294x.2000.01020.x
Cui ZJ, Gao QZ, Liu GN, Pan BT, Chen HL (1996) Planation surfaces, palaeokarst and uplift of Xizang (Tibet) Plateau. Sci China, Ser D Earth Sci 39:391–400
Cunningham CW (1997a) Can three incongruence tests predict when data should be combined? Molec Biol Evol 14:733–740. doi:10.1093/oxfordjournals.molbev.a025813
Cunningham CW (1997b) Is congruence between data partitions a reliable predictor of phylogenetic accuracy? Empirically testing an iterative procedure for choosing among phylogenetic methods. Syst Biol 46:464–478. doi:10.1093/sysbio/46.3.464
Deng JB, Drew BT, Mavrodiev EV, Gitzendanner MA, Soltis PS, Soltis DE (2015) Phylogeny, divergence times, and historical biogeography of the angiosperm family Saxifragaceae. Molec Biol Evol 83:86–98
Doyle JJ, Doyle JL, Rausher J, Brown AHD (2004) Diploid and polyploid reticulate evolution throughout the history of the perennial soybeans (Glycine subgenus Glycine). New Phytol 161:121–132. doi:10.1046/j.1469-8137.2003.00949.x
Drummond AJ, Ho SYW, Phillips MJ, Rambaut A (2006) Relaxed phylogenetics and dating with confidence. PLoS Biol 4:e88. doi:10.1371/journal.pbio.0040088
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molec Biol Evol 29:1969–1973. doi:10.1093/molbev/mss075
Dufresne F, Stift M, Vergilino R, Mable BK (2014) Recent progress and challenges in population genetics of polyploid organisms: an overview of current state-of-the-art molecular and statistical tools. Molec Ecol 23:40–69. doi:10.1111/mec.12581
Escudero M, Hipp AL, Waterway MJ, Valente LM (2012) Diversification rates and chromosome evolution in the most diverse angiosperm genus of the temperate zone (Carex, Cyperaceae). Molec Phylogen Evol 63:650–655. doi:10.1016/j.ympev.2012.02.005
Estep MC, McKain MR, Vela Diaz D, Zhong JS, Hodge JG, Hodkinson TR et al (2014) Allopolyploidy, diversification, and the Miocene grassland expansion. Proc Natl Acad Sci USA 111:15149–15154. doi:10.1073/pnas.1404177111
Farris JS, Källersjö M, Kluge AG, Bult C (1995) Testing significance of incongruence. Cladistics 10:315–319. doi:10.1111/j.1096-0031.1994.tb00181.x
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791. doi:10.2307/2408678
Fortune PM, Pourtau N, Viron N, Aïnouche ML (2008) Molecular phylogeny and reticulate origins of the polyploid Bromus species from section Genea (Poaceae). Amer J Bot 95:454–464. doi:10.3732/ajb.95.4.454
Gao Q (2009) Taxonomic revision of Aconitum L. subgenus Lycoctonum (DC.) Peterm. (Ranunculaceae) from China. PhD Thesis, Institute of Botany, The Chinese Academy of Sciences, Beijing
Gao Q, Ren C, Yang QE (2012) Taxonomic status and distributional range of Aconitum angustius (Ranunculaceae) based on cytological evidence. Nordic J Bot 30:1–13. doi:10.1111/j.1756-1051.2012.01506.x
Ge XJ, Zhang LB, Yuan YM, Hao G, Chiang TY (2005) Strong genetic differentiation of the East-Himalayan Megacodon stylophorus (Gentianaceae) detected by inter-simple sequence repeats (ISSR). Biodivers Conservation 14:849–861. doi:10.1007/s10531-004-0655-6
Grant V (1971) Plant speciation. Columbia University Press, New York
Grant V (1981) Plant speciation, 2nd edn. Columbia University Press, New York
Gregory WC (1941) Phylogenetic and cytological studies in the Ranunculaceae Juss. Trans Amer Philos Soc 31:443–521. doi:10.2307/1005611
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41:95–98
Harmon L, Weir J, Brock C, Glor R, Challenger W (2008) GEIGER: investigating evolutionary radiations. Bioinformatics 24:129–131. doi:10.1093/bioinformatics/btm538
Harrison TM, Copeland P, Kidd WSF, Yin A (1992) Raising Tibet. Science 255:1663–1670. doi:10.1126/science.255.5052.1663
Harrison SP, Yu G, Takahar H, Prentice IC (2001) Palaeovegetation (communications arising): diversity of temperate plants in east Asia. Nature 413:129–130. doi:10.1038/35093166
Hong Y, Gao Q, Luo Y, Luo JP, Zhang Y, Yuan Q, Yang QE (2016) Karyology of Aconitum subgenus Lycoctonum (Ranunculaceae) from China, with a report of the new base chromosome number x = 6 in the genus Aconitum. Nordic J Bot 34:442–454. doi:10.1111/njb.00957
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755. doi:10.1093/bioinformatics/17.8.754
Huelsenbeck JP, Bull JJ, Cunningham CW (1996) Combining data in phylogenetic analysis. Trends Ecol Evol 11:152–158. doi:10.1016/0169-5347(96)10006-9
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Molec Biol Evol 23:254–267. doi:10.1093/molbev/msj030
Jabbour F, Renner SS (2011) Consolida and Aconitella are an annual clade of Delphinium (Ranunculaceae) that diversified in the Mediterranean basin and the Irano-Turanian region. Taxon 60:1029–1040
Jabbour F, Renner SS (2012) A phylogeny of Delphinieae (Ranunculaceae) shows that Aconitum is nested within Delphinium and that Late Miocene transitions to long life cycles in the Himalayas and Southwest China coincide with bursts in diversification. Molec Phylogen Evol 62:928–942. doi:10.1016/j.ympev.2011.12.005
Jakobsson M, Haggenblad J, Tavaré S, Sāll T, Halldén C, Lind-Halldén C et al (2006) A unique recent origin of the allotetraploid species Arabidopsis suecica: evidence from nuclear DNA markers. Molec Biol Evol 23:1217–1231. doi:10.1093/molbev/msk006
Katoh K, Frith MC (2012) Adding unaligned sequences into an existing alignment using MAFFT and LAST. Bioinformatics 28:3144–3146. doi:10.1093/bioinformatics/bts578
Kelly LJ, Leitch AR, Clarkson JJ, Knapp S, Chase MW (2013) Reconstructing the complex evolutionary origin of wild alloploid Tobaccos (Nicotiana section Suaveolentes). Evolution 67:80–94. doi:10.1111/j.1558-5646.2012.01748.x
Kim ST, Donoghue MJ (2008) Incongruence between cpDNA and nrITS trees indicates extensive hybridization within Eupersicaria (Polygonaceae). Amer J Bot 95:1122–1135. doi:10.3732/ajb.0700008
Kreft H, Sommer JH, Barthlott W (2006) The significance of geographic range size for spatial diversity patterns in Neotropical palms. Ecography 29:21–30. doi:10.1111/j.2005.0906-7590.04203.x
Kurita M (1957) Chromosomes studies in Ranuculaceae V. Karyotypes of the subtribe Delphiniinae. Rep Biol Inst Ehime Univ 3:1–8
Langlet O (1927) Beiträge zur Zytologie der Ranuncula. Svensk Bot Tidskr 21:1–17
Levin DA (2002) The role of chromosomal change in plant evolution. Oxford University Press, New York
Li XW, Li J (1993) A preliminary floristic study on the seed plants from the region of Hengduan Mountain. Acta Bot Yunnan 15:217–231
Li JJ, Wen SX, Zhang QS, Wang FB, Zheng BX, Li BY (1979) A discussion on the period, amplitude and type of the uplift of the Qinghai–Xizang (Tibetan) Plateau. Sci Sin 22:1314–1328
Li JJ, Fang XM, Ma HZ, Zhu JJ, Pan BT, Chen HL (1996) Geomorphological and environmental evolution in the upper reaches of the Yellow River during the late Cenozoic. Sci China, Ser D Earth Sci 39:380–390
Liang X, Chen L, Song L, Fei W, He M, He C, Yin Z (2017) Diterpenoid alkaloids from the root of Aconitum sinchiangense W. T. Wang with their antitumor and antibacterial activities. Nat Prod Res 11:1–8. doi:10.1080/14786419.2016.1272113
Lihova J, Shimizu KK, Marhold K (2006) Allopolyploid origin of Cardamine asarifolia (Brassicaceae): incongruence between plastid and nuclear ribosomal DNA sequences solved by a single-copy nuclear gene. Molec Phylogen Evol 39:759–786. doi:10.1016/j.ympev.2006.01.027
Liu K, Warnow TJ, Holder MT, Nelesen S, Yu J, Stamatakis A et al (2012) SATé-II: very fast and accurate simultaneous estimation of multiple sequence alignments and phylogenetic trees. Syst Biol 61:90–106. doi:10.1093/sysbio/syr095
Liu J, Möller M, Provan J, Gao LM, Poudel RC, Li DZ (2013) Geological and ecological factors drive cryptic speciation of yews in a biodiversity hotspot. New Phytol 199:1093–1108. doi:10.1111/nph.12336
Luo Y, Yang QE (2005) Taxonomic revision of Aconitum (Ranunculaceae) from Sichuan, China. Acta Phytotax Sin 43:289–386. doi:10.1360/aps040102
Magallón S, Sanderson MJ (2001) Absolute diversification rates in angiosperm clades. Evolution 55:1762–1780. doi:10.1111/j.0014-3820.2001.tb00826.x
Mansion G, Zeltner L, Bretagnolle F (2005) Phylogenetic patterns and polyploid evolution within the Mediterranean genus Centaurium (Gentianaceae-Chironieae). Taxon 54:931–950. doi:10.2307/25065479
Mavrodiev EV, Chester M, Suárez-Santiago VN, Visger CJ, Rodriguez R, Susanna A et al (2015) Multiple origins and chromosomal novelty in the allotetraploid Tragopogon castellanus (Asteraceae). New Phytol 206:1172–1183. doi:10.1111/nph.13227
Mayrose I, Zhan SH, Rothfels CJ, Magnuson-Ford K, Barker MS, Rieseberg LH et al (2011) Recently formed polyploid plants diversify at lower rates. Science 333:1257. doi:10.1126/science.1207205
Myers N, Mittermeier R, Mittermeier GC, da Fonseca GAB, Kent J (2000) Biodiversity hotspots for conservation priorities. Nature 403:853–858. doi:10.1038/35002501
Oskars TR, Bouchetb P, Malaquiasa MAE (2015) A new phylogeny of the Cephalaspidea (Gastropoda: Heterobranchia) based on expanded taxon sampling and gene markers. Molec Phylogen Evol 89:130–150. doi:10.1016/j.ympev.2015.04.011
Otto SP (2007) The evolutionary consequences of polyploidy. Cell 131:452–462. doi:10.1016/j.cell.2007.10.022
Otto SP, Whitton J (2000) Polyploid incidence and evolution. Annual Rev Genet 34:401–437. doi:10.1146/annurev.genet.34.1.401
Paradis E, Claude J, Strimmer K (2004) APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20:289–290. doi:10.1093/bioinformatics/btg412
Paun O, Fay MF, Soltis DE, Chase MW (2007) Genetic and epigenetic alterations after hybridization and genome doubling. Taxon 56:649–656. doi:10.2307/25065850
Pigg KB, DeVore ML (2005) Palaeoactaea gen. nov. (Ranunculaceae) fruits from the Paleogene of North Dakota and the London clay. Amer J Bot 92:1650–1659. doi:10.3732/ajb.92.10.1650
Plummer M, Best N, Cowles K, Vines K (2006) CODA: convergence diagnosis and output analysis for MCMC. R News 6:7–11
Posada D, Crandall KA (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14:817–818. doi:10.1093/bioinformatics/14.9.817
Qiu YX, Fu CX, Comes HP (2011) Plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Molec Phylogen Evol 59:225–244. doi:10.1016/j.ympev.2011.01.012
Rabosky DL (2014) Automatic detection of key innovations, rate shifts, and diversity-dependence on phylogenetic trees. PLoS ONE 9:e89543. doi:10.1371/journal.pone.0089543
Rabosky DL, Grundler M, Anderson C, Title P, Shi JJ, Brown JW et al (2014) BAMMtools: an R package for the analysis of evolutionary dynamics on phylogenetic trees. Methods Ecol Evol 5:701–707. doi:10.1111/2041-210X.12199
Rambaut A, Suchard MA, Drummond AJ (2013) Tracer v1.6. Available at: http://tree.bio.ed.ac.uk/software/tracer/
Ramsey J, Schemske DW (1998) Pathways, mechanisms, and rates of polyploid formation in flowering plants. Annual Rev Ecol Syst 29:467–501. doi:10.1146/annurev.ecolsys.29.1.467
Ramsey J, Schemske DW (2002) Neopolyploidy in flowering plants. Annual Rev Ecol Syst 33:589–639. doi:10.1146/annurev.ecolsys.33.010802.150437
Ronquist F (1997) Dispersal-vicariance analysis: a new approach to the quantification of historical biogeography. Syst Biol 46:195–203
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574. doi:10.1093/bioinformatics/btg180
Sang T, Crawford DJ, Stuessy TF (1997) Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). Amer J Bot 84:1120–1136. doi:10.2307/2446155
Sauquet H, Ho SY, Gandolfo MA, Jordan GJ, Wilf P, Cantrill DJ et al (2012) Testing the impact of calibration on molecular divergence times using a fossil-rich group: the case of Nothofagus (Fagales). Syst Biol 61:289–313. doi:10.1093/sysbio/syr116
Schilling EE, Panero JL, Crozier BS, Scott RW, Dávila P (2015) Bricklebush (Brickellia) phylogeny reveals dimensions of the great Asteraceae radiation in Mexico. Molec Phylogen Evol 85:161–170. doi:10.1016/j.ympev.2015.02.007
Semenov AA, Enikeev AG, Snetkova LV, Permyakov AV, Sokolova NA, Dudareva LV (2016) Ortho-phthalic acid esters in lipophilic extract from the cell culture of Aconitum baicalense Turcz ex Rapaics 1907. Dokl Biochem Biophys 471:421–422. doi:10.1134/S1607672916060120
Shang XM, Lee CL (1984) Chromosome studies of ten species of Aconitum in China. Acta Phytotax Sin 22:378–385
Sharbel TF, Mitchell-Olds T (2001) Recurrent polyploid origins and chloroplast phylogeography in the Arabis holboellii complex (Brassicaceae). Heredity 87:59–68. doi:10.1046/j.1365-2540.2001.00908.x
Shi YF, Li JJ, Li BY (1998) Uplift and environmental changes of Qinghai–Tibetan Plateau in the late Cenozoic. Guangdong Science and Technology Press, Guangzhou
Silvestro D, Michalak I (2012) RAxMLGUI: a graphical front-end for RAxML. Organisms Diversity Evol 12:335–337. doi:10.1007/s13127-011-0056-0
Slotte T, Ceplitis A, Neuffer B, Hurka H, Lascoux M (2006) Intrageneric phylogeny of Capsella (Brassicaceae) and the origin of the tetraploid C. bursa-pastoris based on chloroplast and nuclear DNA sequences. Amer J Bot 93:1714–1724. doi:10.3732/ajb.93.11.1714
Soltis DE, Soltis PS (1999) Polyploidy: recurrent formation and genome evolution. Trends Ecol Evol 14:348–352. doi:10.1016/S0169-5347(99)01638-9
Soltis DE, Soltis PS, Pires JC, Kovarik A, Tate J (2004) Recent and recurrent polyploidy in Tragopogon (Asteraceae): cytogenetic, genomic and genetic comparisons. Biol J Linn Soc 82:485–501. doi:10.1111/j.1095-8312.2004.00335.x
Soltis DE, Bell CD, Kim S, Soltis PS (2008) Origin and early evolution of angiosperms. Ann New York Acad Sci 1133:3–25. doi:10.1196/annals.1438.005
Soltis DE, Albert VA, Leebens-Mack J, Bell CD, Paterson AH, Zheng C et al (2009) Polyploidy and angiosperm diversification. Amer J Bot 96:336–348. doi:10.3732/ajb.0800079
Song BH, Wang XQ, Wang XR, Sun LJ, Hong DY, Peng PH (2002) Maternal lineages of Pinus densata, a diploid hybrid. Molec Ecol 11:1057–1063. doi:10.1046/j.1365-294X.2002.01502.x
Spicer RA, Harris NB, Widdowson WM, Herman AB, Guo S, Valdes PJ et al (2003) Constant elevation of southern Tibet over the past 15 million years. Nature 421:622–624. doi:10.1038/nature01356
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. doi:10.1093/bioinformatics/btl446
Stebbins GL (1950) Variation and evolution in plants. Columbia University Press, New York
Stebbins GL (1971) Chromosomal evolution in higher plants. Addison-Wesley, London
Sun XJ, Song CQ, Chen XD (1999) China quaternary pollen database (CPD) and Biome 6000 project. Advance Earth Sci 8:407–411
Sun G, Dilcher DL, Wang HS, Chen ZD (2011) A eudicot from the Early Cretaceous of China. Nature 471:625–628. doi:10.1038/nature09811
Swofford DL (2002) PAUP*: phylogenetic analysis using parsimony (*and other methods). Version 4.0 b10. Sinauer Associates, Sunderland
Tamura M (1995) Ranunculaceae. In: Hiepko P (ed) Die Natürlichen Pflanzenfamilien, Aufl. II. 17a (4). Duncker and Humblot, Berlin, pp 223–555
Tamura M, Lauener LA (1979) A synopsis of Aconitum subgenus Lycoctonum: II. Notes Roy Bot Gard Edinburgh 37:431–466
Thompson JD, Lumaret R (1992) The evolutionary dynamics of polyploid plants: origins, establishment and persistence. Trends Ecol Evol 7:302–307. doi:10.1016/0169-5347(92)90228-4
Tuomisto H, Poulsen AD, Ruokolainen K, Moran RC, Quintana C, Celi J, Cañas G (2003) Linking floristic patterns with soil heterogeneity and satellite imagery in Ecuadorian Amazonia. Ecol Applic 13:352–371. doi:10.1890/1051-0761(2003)013[0352:LFPWSH]2.0.CO;2
Utelli AB, Roy BA, Baltisberger M (2000) Molecular and morphological analyses of European Aconitum species (Ranunculaceae). Pl Syst Evol 224:195–212. doi:10.1007/BF00986343
Verboom GA, Bergh NG, Haiden SA, Hoffmann V, Britton MN (2015) Topography as a driver of diversification in the Cape Floristic Region of South Africa. New Phytol 207:368–376. doi:10.1111/nph.13342
Wada K, Takeda K, Haraguchi M, Abe Y, Kuwahara N, Suzuki S, Terui A, Masaka T, Munakata N, Uchida M, Nunokawa M, Yamashita H, Goto M, Lee KH (2016) Four new diterpenoid alkaloids from Aconitum japonicum. Pl Med (Stuttgart) 81:S1–S381
Wang W, Lu AM, Ren Y, Endress ME, Chen ZD (2009) Phylogeny and classification of Ranunculales: evidence from four molecular loci and morphological data. Perspect Pl Ecol Evol Syst 11:81–110. doi:10.1016/j.ppees.2009.01.001
Wang H, Qiong L, Sun K, Lu F, Wang YG, Song ZP et al (2010) Phylogeographic structure of Hippophae tibetana (Elaeagnaceae) highlights the highest microrefugia and the rapid uplift of the Qinghai–Tibetan Plateau. Molec Ecol 19:2964–2979. doi:10.1111/j.1365-294X.2010.04729.x
Wang JF, Pan YZ, Gong X, Chiang CY, Kuroda C (2011) Chloroplast DNA variation and phylogeography of Ligularia tongolensis (Asteraceae), a species endemic to the Hengduan Mountains region of China. J Syst Evol 49:108–119. doi:10.1111/j.1759-6831.2011.00117.x
Wang W, Liu Y, Yu SX, Gao TG, Chen ZD (2013) Gymnaconitum, a new genus of Ranunculaceae endemic to the Qinghai–Tibetan Plateau. Taxon 62:713–722. doi:10.12705/624.10
Weeks A, Simpson BB (2004) Molecular genetic evidence for interspecific hybridization among endemic Hispaniolan Bursera (Burseraceae). Amer J Bot 91:976–984. doi:10.3732/ajb.91.6.976
Wen J, Zhang JQ, Nie ZL, Zhong Y, Sun H (2014) Evolutionary diversifications of plants on the Qinghai–Tibetan Plateau. Frontiers Genet 5:4. doi:10.3389/fgene.2014.00004
Wendel JF, Doyle JJ (2004) Polyploidy and evolution in plants. In: Henry R (ed) Diversity and evolution in plants. CABI Publishing, Wallingford, pp 97–117
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis M, Gelfand D, Sninsky J, White T (eds) PCR Protocols: a guide to methods and applications. Academic Press, San Diego, pp 315–322
Wood TE, Takebayashi N, Barker MS, Mayrose I, Greenspoon PB, Rieseberg LH (2009) The frequency of polyploid speciation in vascular plants. Proc Natl Acad Sci USA 106:13875–13879. doi:10.1073/pnas.0811575106
Woolley SM, Posada D, Crandall KA (2008) A comparison of phylogenetic network methods using computer simulation. PLoS ONE 3:e1913. doi:10.1371/journal.pone.0001913
Wu ZY (1988) Hengduan mountain flora and her significance. J Japan Bot 63:1–15
Xu TT, Abbott RJ, Milne RI, Mao K, Du FK, Wu GL, Ciren ZX, Miehe G, Liu JQ (2010) Phylogeography and allopatric divergence of cypress species (Cupressus L.) in the Qinghai–Tibetan Plateau and adjacent regions. BMC Evol Biol 10:194. doi:10.1186/1471-2148-10-194
Yang QE (1996) A karyotype study of 15 species in the tribe Delphineae (Ranunculaceae) from China. Acta Phytotax Sin 34:39–47
Yang QE (1999) Taxonomic notes on some species of Aconitum L. (Ranunculaceae) from Yunnan, China. Acta Phytotax Sin 37:545–590
Yang QE (2001) Cytology of 12 species in Aconitum L. and of 18 species in Delphinium L. of the tribe Delphineae (Ranunculaceae) from China. Acta Phytotax Sin 39:502–514
Yang QE, Gu ZJ, Wu ZY (1994) A karyomorphological study in Aconitum subgen. Lycoctonum (Ranunculaceae) from Yunnan. Acta Botanica Yunnanica 16:61–74
Yi Z, Strüder-Kypke M, Hu X, Lin X, Song W (2014) Sampling strategies for improving tree accuracy and phylogenetic analyses: a case study in ciliate protists, with notes on the genus Paramecium. Molec Phylogen Evol 71:142–148. doi:10.1016/j.ympev.2013.11.013
Yost JM, Bontrager M, McCabe SW, Burton D, Simpson MG, Kay KM, Ritter M (2013) Phylogenetic Relationships and Evolution in Dudleya (Crassulaceae). Syst Bot 38:1096–1104. doi:10.1600/036364413X674760
Yu Y, Harris AJ, Blair C, He XJ (2015) RASP (Reconstruct Ancestral State in Phylogenies): a tool for historical biogeography. Molec Phylogen Evol 87:46–49. doi:10.1016/j.ympev.2015.03.008
Yuan YW, Olmstead RG (2008) A species-level phylogenetic study of the Verbena complex (Verbenaceae) indicates two independent intergeneric chloroplast transfers. Molec Phylogen Evol 48:23–33. doi:10.1016/j.ympev.2008.04.004
Yuan Q, Yang QE (2006) Polyploidy in Aconitum subgenus Lycoctonum (Ranunculaceae). Bot J Linn Soc 150:343–353. doi:10.1111/j.1095-8339.2006.00468.x
Zhao Y, Bu G, Zhou Y, Lv L, Yan G, Chen B, Wang L, Cen X (2010) Mechanism study of Aconitum-induced neurotoxicity in PC12 cells: involvement of dopamine release and oxidative damage. Neurotoxicology 31:752–757. doi:10.1016/j.neuro.2010.06.005
Acknowledgements
We would like to thank Prof. Youngdong Kim for providing the samples of Aconitum longecassidatum, A. pseudolaeve and A. quelpaertense from Korea, and Dr. Tetsuo Ohi-Toma for providing the sample of A. pterocaule from Japan. Special thanks to Zheng Li and Anthony Baniaga for helpful suggestions and discussion on the data analyses. Dr. A.J. Harris is thanked for the valuable suggestions on the manuscript.
Sources of funding
This work was supported by the Natural Science Foundation of China (Project No. 31470319), the Main Direction Program of Knowledge Innovation of the Chinese Academy of Sciences (KSCX2-EW-J-28), and the Natural Science Foundation of Guangdong Province, China (Project No. 10151065005000019).
Sample
No specific permits were required for the species of Aconitum subgenus Lycoctonum used in the current study, and all samples were collected by researchers following current Chinese regulations.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling editor: Hiroshi Kudoh.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Information on Electronic supplementary material
Information on Electronic supplementary material
Online Resource 1. List of species, code number, voucher information, chromosome number (ploidy level), geographical and DIVA regions for subgenus Lycoctonum species.
Online Resource 2. The best-scoring maximum likelihood (ML) tree inferred from cpDNA data set with the GTRGAMMA model implemented in RAxML.
Online Resource 3. The best-scoring maximum likelihood (ML) tree inferred from nrDNA data sets with the GTRGAMMA model implemented in RAxML.
Online Resource 4. Filtered supernetwork constructed from separate cpDNA and nrDNA gene trees.
Rights and permissions
About this article

Cite this article
Kong, H., Zhang, Y., Hong, Y. et al. Multilocus phylogenetic reconstruction informing polyploid relationships of Aconitum subgenus Lycoctonum (Ranunculaceae) in China. Plant Syst Evol 303, 727–744 (2017). https://doi.org/10.1007/s00606-017-1406-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00606-017-1406-y