
Abstract
Chlorophyllase (Chlase) was one of first plant enzymes to be characterized biochemically, but its phylogenetic position and tertiary structure remain unknown. In this study, two isoforms of the Chlase gene, PmCLH1 and PmCLH2, were cloned from Pachira macrocarpa (Pm), and they shared 84 % identity in amino acid sequences. An unrooted phylogenetic tree classified 38 selected PmCLH homologous sequences into four clades, and PmCLHs were placed in the same clade as Arabidopsis AtCLH1. Moreover, the recombinant PmCLHs exhibited chlorophyll degradation activity in vitro. Tertiary structure modeling revealed that the catalytic triad of PmCLHs and AtCLHs were located on surface-exposed loops and clustered as a catalytic domain. Expressions of PmCLHs were not transcriptionally active in all tissues, and PmCLH1 and PmCLH2 were more abundant in young and old leaves, respectively. Multi-band patterns were observed in both young and old leaves of P. macrocarpa according to a Western blot analysis, suggesting that posttranslational modification was required for PmCLH maturation. In conclusion, we present the first report on the evolution and tertiary structural modeling of Chlase. We also provide evidence that mRNA and protein levels of PmCLHs are unequally expressed in young and old leaves of P. macrocarpa.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chlorophyll (Chl) is the most abundant photosynthetic pigment in higher plants, and is bound to Chl a/b-binding proteins, cytochrome b6 and early-light-inducible proteins embedded in thylakoid membranes of chloroplasts for their functions in photosynthesis (Bruno and Wetzel 2004). More than one Chl degradation pathway has been elucidated upon different physiological and developmental conditions, including chlorophyllase (Chlase) degrading Chl to phytol and chlorophyllide (Chlide), leading to a rate-limiting step in the Chl-degradation pathway (Harpaz-Saad et al. 2007). In addition, pheophytinase (PPH), which is responsible for converting Mg2+-free Chl to pheophorbide (pheide), may lead to another Chl degradation pathway in senescing leaves (Schelbert et al. 2009). Both proposed pathways show that Chl is cleaved into several intermediate catabolites in chloroplasts. Although biological functions of Chlase in Chl degradation and cell damage were reported (Kariola et al. 2005), only a few identified Chlases were proven to be localized in chloroplasts. For example, two Chlases identified from two woody plants, citrus and ginkgo, were demonstrated to directly transit peptides from their chloroplast target (Jacob-Wilk et al. 1999; Okazawa et al. 2006), and Chlase activity in the herbaceous plant, barley, was detected in chloroplasts (Matile et al. 1997). In Arabidopsis, the AtCLH isoform, located in chloroplasts, was experimentally demonstrated (Schenk et al. 2007). To better understand the molecular and physiological basis of Chlase in Chl degradation, structural elucidation and functional characterization of Chlase from diverse species should be done.
Many Chlase genes were cloned and annotated from diverse species and genome projects. Comparison of deduced amino acid sequences of Chlase reveals that the lipase motif and catalytic triad are highly conserved among all known Chlases isolated from different species. However, the homology of whole amino acid sequences among these Chlases is low (Tsuchiya et al. 1999; Takamiya et al. 2000). Integration of these data trends implies that Chlase gene evolution, especially sequences encoding the N-terminal transit peptide domain, may play a role in its physiological function and ubcellular location. Furthermore, growing evidence indicates that Chlase may function as a dimeric protein in vivo (Nishiyama et al. 1994), and its enzymatic properties in vitro varied among species (Hiba and Selim 1997; Yang et al. 2004; Arkus et al. 2005; Yunyu et al. 2006). The important catalytic triad on the primary structure of Chlase was completely identified, but topological features of active sites on the tertiary structure remain unclear. To reveal the diverse enzymatic properties of Chlase, additional efforts to elucidate the subsequent tertiary structure are needed.
No information is available regarding the biological function of Chlase in evergreen plants. We thus compared Chlase activities of seven selected evergreen plants and found that P. macrocarpa contained significantly higher Chlase activities than the others, and the activities were localized in the subcellular fraction of the chloroplast envelope (unpublished data). Accordingly, two full-length genes of Chlase isoforms were cloned from P. macrocarpa, which share a high degree of identity throughout the entire amino acid sequences, belong to the same clade, and exhibit Chlase activities in vitro. The tertiary structure modeling of Chlase demonstrated the catalytic triad on surface-exposed loops and a cluster as a catalytic domain. The differential expression patterns of the two gene and protein levels of the Chlase isoforms from various tissues suggest that these two isoforms have different biological functions in vivo.
Materials and methods
Plant material and preparation of acetone powder
Pachira macrocarpa was purchased from local gardens; the liquid-nitrogen-frozen leaves were ground up with a mortar and pestle and then homogenized with pre-chilled acetone (−20 °C). After centrifuging the homogenate at 3,000g and 4 °C for 5 min, the precipitate was collected. The cold acetone extraction was repeated, and the sample was homogenized several times to remove all traces of chls and carotenoids. The acetone powder was dried with nitrogen and stored at –20 °C until use.
Assay of Chlase activity
To determine Chlase activity, 100 mg of acetone powder was homogenized with 5 ml extraction buffer, containing 5 mM potassium phosphate (pH 7.0), 50 mM KCl, and 0.24 % Triton X-100 for 1 h at 30 °C. The supernatant, after centrifuging at 15,000g for 15 min, was used for the enzyme assay. The assay of Chase activity followed a modified method of McFeeters et al. (1971). The standard reaction mixture contained 0.1 ml of substrate (1 μmol/ml chl a or b), 0.1 ml of the above supernatant, and 0.8 ml of reaction buffer containing 100 mM sodium phosphate (pH 7.0), and 0.24 % Triton X-100. The mixture was incubated for 60 min at 30 °C, and the reaction was stopped with 1.0 ml of 10 mM KOH. After reacting, 1 ml of the mixture was further mixed with 5 ml of a hexane/acetone (3:2, v/v) solvent to eliminate the interference of chl. The product, chlorophyllide a or b, in the acetone phase was determined by a Hitachi U-2000 spectrophotometer using an extinction coefficient of 74.9 mM−1 cm−1 at 667 nm or 47.2 mM−1 cm−1 at 650 nm for chlorophyllide a or b, respectively (Trebitsh et al. 1993). One unit of Chlase a or b degradation activity was defined as the amount of enzyme needed to catalyze the production of 1 μmol of chlorophyllide a or b per minute.
Statistical analysis
Different plants as replicates were calculated to determine the average Chlase activity, and a completely randomized design with at least three replications was used. Data were analyzed by SAS 8.2 (SAS Institute, Cary, NC, USA). For significance levels, means of the Chlase a, b, and a+b activities of the seven evergreen plants were separated by the least significant difference (LSD) at p ≤ 0.05. Moreover, means of the Chlase a, b, and a+b activities between the envelope and thylakoid membrane were compared using an unpaired t test at a 0.05 probability level.
RNA isolation
Total RNA was isolated from 1 g of shoots, young leaves, and old leaves of P. macrocarpa with a Qiagen RNeasy plant Mini Kit (Valencia, CA, USA), and then poly (A)+ mRNA was extracted from the total RNA with a Qiagen Oligotex Mini Kit according to the vendor’s instructions. Concentrations of total RNA and messenger (m)RNA were determined by a NanoDrop ND-1000 spectrophotometer (Paris, France) at 260 nm.
Real-time reverse-transcription quantitative polymerase chain reaction (RT-qPCR) and quantification of RNA levels
RT-qPCR analysis was employed to quantify and validate relative changes in the expressions of Chlase genes. Total RNA of P. macrocarpa from shoots, young leaves, and old leaves was isolated as described above. The first-strand complementary (c) DNA synthesis was performed with an Oligo (dT) primer using RETROscript Reverse Transcription for the RT-PCR kit (Ambion, Austin, TX, USA). The cDNA was diluted 1:5 for the real-time PCRs which were carried out in 384-well plates in a Light Cycler 480 (Roche, Basel, Switzerland). The gene-specific primers for PmCLH1 (1F: 5′-TCAACGAGTGCAAACCTCCG-3′ and 1R: 5′-AGCCCATAATCCATATATGCTC-3′) and PmCLH2 (2F: 5′-GAGTCCAAGGGCTGCATGC-3′ and 2R: 5′-CTAATGTAACCTTTGTACGG-3′) were used in the RT-qPCR. The correct size of the amplified region for each primer was checked by agarose gel electrophoresis. Each RT-qPCR (20 μl) contained 1 μl of the diluted cDNA, 9 μl of water, 2 μl of primer mix (10×), 4 μl of 5× SYBR green I, 1.6 μl MgCl2, 0.4 μl of the enzyme mix, and 2 μl of resolution solution (Roche RNA Amplification Kit). The amplification program consisted of one cycle at 95 °C for 5 min for pre-incubation, followed by 40 cycles at 95 °C for 30 s, 58 °C for 10 s, and 72 °C for 20 s. After amplification, a melting-curve analysis was run using the program of one cycle at 95 °C for 5 s, 65 °C for 10 s, and 95 °C with 0 s held in the step-acquisition mode, followed by cooling at 40 °C for 30 s. Three replicates were performed for each cDNA sample, and template-free and negative controls were used. To normalize the total amount of cDNA in each reaction, the Rubisco gene of P. macrocarpa (unpublished data) was co-amplified as the internal control using primers of RubiscoF (5′-GTGGTGGTTCAATCATGTTCCC-3′) and RubiscoR (5′-TTTCCTTGCTCATTCGGTCTG-3′). Data were analyzed with the PCR efficiency correction using Light Cycler 480 Relative Quantification software V1.01 (Roche) based on relative standard curves describing the PCR efficiencies of the Chlase and reference genes. These relative RNA quantities of RNA samples are presented as “expression” values in Table 2, and these values allowed comparisons of relative RNA amounts among tissues.
Molecular cloning of full-length genes of PmCLHs
To obtain partial sequences of Chlase genes from P. macrocarpa, a pair of degenerate primers (CLH5: 5′-TTCDCATGGHTWCATHSTYRTHGCYCC-3′and CLH3: 5′-TCRTCHARNAWRTCMRWRTgNCC RTA-3′) was designed based on highly conserved amino acid sequences of Chlase. Total RNA was extracted from leaves of P. macrocarpa using an RNeasy Mini Kit (Qiagen, Hilden, Germany). An aliquot of 1 μg total RNA was reverse-transcribed into cDNA with an oligo d(T)18 primer. PCR fragments were subcloned into a pGEM-T Easy vector (Promega, Madison, WI, USA), and sequences were verified by sequencing. To clone full-length Chlase genes from P. macrocarpa, a 5′ and 3′ RACE (rapid amplification of cDNA ends) was conducted according to the manufacturer’s instructions. Primers used in the 5′ and 3′ RACE (Pm1F: 5′-ATCGACCCATGTTGCTGGG-3′, Pm1R: 5′-TCAAAGTTGCCTAACGTAATCT-3′, Pm2F: 5′-GAGTCCAAGGGCTGCATGC-3′, and Pm2R: 5′-CAAATAATCTTCCCTCACCT-3′) were designed based on cloned partial Chlase genes. PCR conditions used in the 3′ and 5′ RACE were the same and were conducted as follows: 94 °C for 3 min followed by 35 cycles of amplification (94 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min), and a final extension at 72 °C for 3 min. All amplicons were subcloned into the pGEM-T Easy vector, and sequences were verified by sequencing.
Expression of PmCLHs in Escherichia coli
Full-length and truncated version of PmCLHs cDNAs were constructed, respectively, in the pET-24a or pET-28a expression vector under the control of the T7 promoter. EcoRI and XhoI sites were added to the primers (PmCLH1, full-length forward primer: 5′-ATATGAATTCATGCCTTCGGTTTTTGTATCC-3′; truncated forward primer: 5′-ATATGAATTCCCTCCTAAGCCACTTTTGATT-3′ and reverse primer: 5′-ATATCTCGAGAAGTTGCCTAACGTAATCTTTG-3′; PmCLH2, full-length forward primer: 5′-ATATGAATTCATGGCACAGCTATTGGAAACT-3′; truncated forward primer: 5′-ATATGAATTCCCTCCTAAGCCACTGTTGATT-3′ and reverse primer: 5′-ATATCTCGAGAACATTGAACTGAACTTGATC-3′) to amplify the inserted fragments. The PCR fragments were digested with EcoRI and XhoI and subsequently ligated to an EcoRI-XhoI-digested pET-24a or pET-28a T7 expression vector (Novagen, Darmstadt, Germany). All constructed plasmid DNA was transformed into E. coli strain BL21 (DE3) for protein overexpression. Cells were cultivated at 37 °C, and the recombinant PmCLHs were induced by the addition of isopropyl thio-β-galactoside (IPTG) to a final concentration of 1 mM. Cells were harvested by centrifugation, and cell pellets were resuspended in extraction buffer (5 mM KH2PO4, 5 mM K2HPO4, 50 mM KCL, and 0.24 % Triton X-100) and sonicated to disrupt the cells. To determine protein concentration, tenfold serial dilution of whole-lysate was prepared. Thus, the interference of Triton X-100 in Bradford assay could be excluded. Aliquots of soluble E. coli total protein were quantified by the Bradford dye-binding assay (Biorad, Hercules, CA) for the following assays and analyzed by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE). Recombinant full-length PmCLHs were overexpressed in E. coli, and total soluble proteins were used for Chlase activity assays. Chl a and b (1 μmol/ml) degradation kinetics of PmCLH1 and PmCLH2 were determined separately.
Antibody preparation and Western blot analysis
Previously, recombinant fusion proteins were used as source of enzyme on Chase activity assay (Beneditti and Arruda 2002; Tsuchiya et al. 2003). In the study, we used the His-tagged PmCLHs in enzymatic activity assay. Hi-tagged PmCLHs were expressed in E. coli, and Chlase activity in the lysates was measured. The lysate of E. coli carrying the empty vector pET28a was used as negative control. To generate specific antibodies for detecting PmCLHs, recombinant PmCLH1 was purified by a nickel-affinity column and then used to immunize rabbits for polyclonal antibody generation. Recombinant PmCLH1 was eluted from SDS-PAGE according to Chuang et al. (1996) to generate polyclonal antibodies in rabbits. Proteins were separated by 12.5 % SDS-PAGE and stained with Coomassie brilliant blue R-250. Proteins separated by SDS-PAGE were transferred to nitrocellulose membranes and blocked with 3 % bovine serum albumin (BSA) (Sigma-Aldrich, Saint Louis, MO, USA). Membranes were than separately hybridized with antibodies including anti-PmCLH1, anti-LC2B, anti-AtTic110, and anti-Toc75. After washing, membranes were probed with an anti-rabbit secondary antibody conjugated with alkaline phosphatase, and then a 5-bromo-4-chloro-3-indolyl phosphate (BCIP)/nitro blue tetrazolium (NBT) (Sigma-Aldrich) signal was developed on the membranes.
Structural analyses
To predict subcellular localization of proteins, the ChloroP 1.1 server (Emanuelsson et al. 1999), TargetP 1.1 server (Emanuelsson et al. 2000), Predotar (Small et al. 2004), and WoLF PSORT (Horton et al. 2007) were used. For secondary and tertiary structure predictions and modeling, the I-TASSER server (Zhang 2008) was used.
Phylogenetic analysis
Phylogenetic analyses of Chlase sequences were preformed with MEGA4 software (Tamura et al. 2007). Thirty-eight Chlase homologous amino acid sequences (Supplemental Table S1) from different species acquired from the NCBI database were aligned by Clustal W (Thompson et al. 1994). The evolutionary history and a bootstrap consensus tree were reconstructed using the Neighbor-joining (NJ) method (Saitou and Nei 1987) from 500 replicates. The optimal tree with the sum of branch lengths (7.74374846) was drawn to scale (Fig. 2). The branch lengths were the same units as those of the evolutionary distances for the phylogenetic tree. All positions containing alignment gaps and missing data were eliminated in the pairwise sequence comparisons (pairwise deletion option), and 340 positions were used.
Results
Molecular cloning and tertiary structural modeling of Chlase from P. macrocarpa
To clone the Chlase gene from P. macrocarpa, a degenerate PCR followed by 5′ and 3′ RACE cloning methods was applied. Two putative Chlase clones, tentatively named PmCLH1 (accession no. FJ754215) and PmCLH2 (accession no. FJ754216), were subcloned and sequenced. Sequencing results revealed that the full-length cDNAs of PmCLH1 and PmCLH2 were, respectively, 1,189 and 1,309 bp long. The polypeptide sequence deduced from the cDNA of PmCLH1 encoded a putative protein of 339 amino acid residues and had a predicted molecular weight of 36.69 kDa. The PmCLH2 gene encoded 314 amino acid residues with a predicted molecular mass of 33.78 kDa. PmCLH1 and PmCLH2 shared 84 % identity for the entire amino acid sequence. Figure 1a demonstrates comparisons of the homologous polypeptide sequences, consensus catalytic triads, and lipase motifs in PmCLHs and selected Chlases. The subcellular localization of PmCLHs was predicted using four web-based prediction programs and showed that PmCLH1 was localized in chloroplasts, whereas PmCLH2 might be a cytoplasmic protein (Table 1). Furthermore, protein structural modeling of PmCLHs was analyzed by the I-TASSER program to locate the active site architecture of Chlase. The model was generated by threading the amino acid sequences of PmCLHs and AtCLHs into the three-dimensional tertiary structure of their closest homologue, Streptomyces exfoliatus lipase (Wei et al. 1998). The sequence identity and similarity between S. exfoliatus lipase and PmCLH1 were 14 and 72 %, respectively, whereas PmCLH2 shared 15 % identity and 78 % similarity with the template from the overall sequence. Obviously, the catalytic triad composed of serine, histidine, and aspartic acid was located on surface-exposed loops and clustered as a catalytic domain (Fig. 1b). Comparison of the tertiary structures of PmCLHs and AtCHLs indicated that the essential features of these four structures were quite similar, especially as to the active site architecture.

Comparative sequence analysis of Pachira macrocarpa chlorophyllases (PmCLHs) from primary sequence alignments to three-dimensional structure modeling. a Multiple amino acid sequence alignments of chlorophyllase (Chlase) from Pachira macrocarpa (PmCLH1 and PmCLH2), Arabidopsis thaliana (AtCLH1 and AtCLH2), Brassica oleracea (BoCLH1), Zea mays (ZmCLH2), Piper betle (PbCLH), and Ginkgo biloba (GbCLH). Conserved amino acids among Chlases are highlighted by dark squares, and similar amino acids are indicated in gray. The N-terminal truncated site of PmCLHs in the recombinant protein is shown by a downward arrow. Predicted secondary structural elements are shown as α-helices and β-strands above the sequences. The catalytic triad and lipase motif are, respectively, indicated by red asterisks and underlining. b Ribbon schematic of the predicted tertiary structures of PmCLHs and AtCLHs. The diagrams were constructed using the I-TASSER structure modeling algorithm. The conserved catalytic triads (serine, aspartic acid, and histidine) of PmCLHs and AtCLHs are indicated
Sequence homology and phylogenetic analysis of PmCLHs
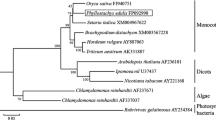
To further classify the phylogenetic position of cloned PmCLHs, an unrooted phylogenetic tree was constructed on the basis of the alignment of PmCLH-homologous sequences from moss, angiosperms, and gymnosperms. The tree was built by the NJ method using full-length amino acid sequences, revealing that 38 selected sequences were divided into four clades (Fig. 2). Sequences from Picea sitchensis, Ginkgo biloba, and Physcomitrella patens ssp. patens were clustered together in clade I, the most ancient clade in this study. The remaining sequences from a wide range of angiosperm species were divided into monocots (clade II) and dicots (clades III and IV), and both PmCLH1 and PmCLH2 were in clade III (Fig. 2). Interestingly, many dicot species contained more than one Chlase homolog, for instance, Arabidopsis thaliana, Glycine max, Brassica oleracea, Vitis vinifera, and Populus trichocarpa, that were classified into different clades.

Phylogenetic analysis of Pachira macrocarpa chlorophyllase (PmCLH) and its homologous proteins. The unrooted phylogenetic tree was constructed in MEGA4 using the Neighbor-joining method with 500 bootstrap replicates of the ClustalW alignment. Evolutionary distances were estimated using the Poisson correction model. Numbers indicate the occurrence of a given branch in 500 bootstrap replicates of the given dataset. The scale bar represents 0.1 amino acid substitutions per site
Recombinant PmCLHs show Chl degradation activities in vitro
Based on the results of the amino acid sequence analysis and phylogenetic classification of PmCLHs, spatial localization and enzymatic activity were examined to evaluate the physiological functions of PmCLHs in plant cells. To eliminate the hydrophobic effect of transit peptide on the Chlase activity assay, if any, we performed the assay with wild type and N-terminal truncated PmCLHs versions based on sequence alignment as shown in Fig. 1 and in the case of GbCLH reported by Okazawa et al. (2006). Thus, four recombinant expression plasmids were constructed and transformed into E. coli to produce recombinant PmCLHs (Fig. 3a). To generate specific antibodies for detecting PmCLHs, recombinant PmCLH1 was purified by a nickel-affinity column, and then used to immunize rabbits for polyclonal antibody generation. According to the results of the Western blot analysis, the predicted molecular mass of each His-tagged PmCLHs fusion protein was detected as a major signal from crude E. coli lysates, along with minor additional bands present in IPTG-induced cell lysates but absent from non-induced BL21 total proteins (Fig. 3b). This may have been caused by partial degradation of the recombinant PmCLHs during expression. Therefore, the in vitro Chlase activity assay was subsequently conducted using IPTG-induced E. coli lysates as the source of the recombinant PmCLHs as well as Chl a and b as substrates. Results show that both the full-length PmCLH1 and PmCLH2 exhibited significantly higher abilities to degrade Chl a and b compared with the truncated PmCLHs (Fig. 4c, d). Moreover, PmCLH1 showed significant higher Chl-degradation activity than PmCLH2 (Fig. 4a, b). The results indicate that Chl b was recognized and degraded with greater efficiency than Chl a in both PmCLH1 and PmCLH2.

Fusion protein construction, expression, and Western blot analysis of Pachira macrocarpa chlorophyllases (PmCLHs). a His-taq fusion constructions of PmCLHs, including both full-length PmCLHs (F-PmCLHs) and truncated PmCLHs (T-PmCLHs), were cloned into pET24a or pET28a expression vectors as indicated. b Recombinant PmCLHs were overexpressed in E. coli strain BL21 induced by 1 mM IPTG. Total protein of 5 μg of E. coli was loaded and resolved in a 12.5 % SDS-PAGE (upper panel). Recombinant PmCLHs using polyclonal antibodies raised against PmCLH1 were analyzed by Western blotting (lower panel)

Characterization of in vitro recombinant Pachira macrocarpa chlorophyllase (PmCLH) activities. Recombinant full-length PmCLHs were overexpressed in E. coli, and total soluble proteins were used for Chlase activity assays. Chl a and b (1 μmol/ml) degradation kinetics of PmCLH1 (a) and PmCLH2 (b) were determined separately. Chlase in vitro activities of PmCLHs were examined in the presence (c) or absence of Triton X-100 (d) in the reaction reagents. Pm1(2)F, PmCLH1(2) full-length protein, Pm1(2)T, PmCLH1(2) N-terminal-truncated protein. N.D. not detected. Values represent the mean ± SD of three independent experiments
Differential mRNA and protein expressions of PmCLHs in various tissues
A quantitative PCR was conducted to assess the mRNA expressions of PmCLHs from shoots and two different maturation stages of leaves. We normalized the expression level of the PmCLH gene using Rubisco transcript levels determined in the same quantitative PCR reaction. The transcription level of PmCLH1 in young leaves was significantly higher than those in old leaves and shoots (Table 2). However, PmCLH2 gene transcription in old leaves was four- and fivefold significantly higher than those of young leaves and shoots, respectively. Moreover, the expression of PmCLH1 was significantly induced in young leaves compared with PmCLH2. No significant differences in old leaves or shoots in response to PmCLH genes were observed. Protein expression patterns of PmCLHs between young and old leaves by the Western blot analysis using polyclonal antibodies raised against PmCLH1 were also compared. Many signals were shown in young leaves consisting of two molecular mass bands at 53 and 75 kDa, one expected molecular mass at 37 kDa, and three molecular weights at between 30 and 34 kDa. In contrast, two strong signals at a molecular mass of 32 kDa and three minor signals spanning molecular masses from 37 to 53 kDa were observed in old leaves (Fig. 5).

Western blot analysis of total proteins extracted from young and old leaves of Pachira macrocarpa, and hybridized to polyclonal antibodies raised against PmCLH1. Samples containing 20 μg of total proteins were loaded into a 12.5 % SDS-PAGE (left panel). The duplicate SDS-PAGE was then transferred to a Nylon membrane and hybridized to polyclonal antibodies raised against PmCLH1 (right panel)
Discussion
In this study, seven evergreen plants were screened for Chlase activity. Pachira macrocarpa showed significantly higher Chlase activity compared with other plants. Two Chlase homologous genes, PmCLH1 and PmCLH2, were cloned from leaves of P. macrocarpa and their mRNA showed differential expression levels in young and old leaves. Both PmCLHs contained predictable transit peptide sequences, and they played roles in Chl degradation activities in vitro. Furthermore, the in vitro enzymatic activity assay also revealed that PmCLHs degraded Chl b more efficiently than Chl a, and thus this may be further support for their close phylogenetic relationship.
The advantage of genetic engineering techniques is in understanding how the function of Chlase in vivo has evolved. AtCLHs (A. thaliana) and Chlase 1 (Citrus sinensis) genes are commonly used in transgenic plants to reveal the function and location of Chlase. Accordingly, it was suggested that Chlase functions as a Chl scavenger and in pathogen defense in plants (Hortensteiner 2006). However, some controversial issues on Chlase activity in maturing fruit and its intracellular localization remain to be resolved (Yamauchi et al. 1997; Distefano et al. 2009). There is no doubt that molecular cloning and functional characterization of Chlase from diverse species are important in elucidating the biological functions of Chlase. In this study, PmCLH genes were cloned from an evergreen plant, P. macrocarpa, and PmCLH in vitro activities were analyzed in greater detail. Based on the q-PCR results, expression patterns of PmCLHs greatly differed in different tissues of P. macrocarpa. Although PmCLH transcripts were detected in all tissues, the PmCLH1 gene was most strongly active and regulated in young leaves of P. macrocarpa (Table 2). Tissue specificity or signal responsiveness of gene expression often reflects the function of the corresponding gene products in plant development and signaling. It is also known that Chl contents in young and old leaves differ, and Chlase plays a role in Chl biosynthesis (Fiedor et al. 1992). Different expression patterns of PmCLHs in different tissues suggest that they may engage in different biological functions. Furthermore, PmCLH proteins expressed in young and old leaves exhibited multiple bands over a wide range of molecular weights on the Western blots (Fig. 5). This may have been caused by posttranslational modification such as glycosylation (Terpstra 1981) or N-terminal processing (Azoulay-Shemer et al. 2008). In addition, Banas et al. (2011) reported that the levels of mRNA of two enzymes were involved in chlorophyll catabolism in Arabidopsis, and products of AtCLH1 and AtCLH2 genes were increased upon illumination with white light. In our study, the involvement of light in the regulation of PmCLH1 and PmCLH2 expression and its relation to senescence were unclear, but the fundamental differences in the tissues of their transcription suggest that these enzymes play different roles in the metabolism of Chl. The enzyme expression is strongly tissue controlled, at least at the level of RNA.
Phylogenetic analysis of 38 selected Chlase isozymes classified them into four major taxonomic clades in this study. Chlase sequences acquired from ancestor species such as moss, ginkgo, and Sitka spruce clustered together in clade I; monocot species including rice, maize, and sorghum formed clade II; and dicot species were further classified into clades III and IV. PmCLHs were grouped together in clade III (Fig. 2). AtCLH1 was also located in this clade, and its gene expression was reported to be upregulated by methyl jasmonate (Tsuchiya et al. 1999). It is known that the function of Chlase is associated with the physiological states of leaf senescence, fruit maturity, and damage control in plants (Kariola et al. 2005; Harpaz-Saad et al. 2007; Azoulay-Shemer et al. 2008). Evidently, in most of the studied flowering plants, more than one isozyme of Chlase exists (Trebitsh et al. 1993; Tsuchiya et al. 1997). The exact physiological significance of each isozyme in the same species remains unclear. The phylogenetic analysis in this study to some extent implies evolutionary and functional constraints of Chlase isozymes. Thus, it may be inferred that clade III Chlase isozymes play vital roles in plant defense.
Several lines of evidence suggested that Chlase is an intrinsic membrane protein and it may contain transit peptides at the N-terminal for chloroplast targeting (Terpstra 1978). Prediction of PmCLH subcellular localization revealed that PmCLHs may target distinct subcellular compartments, in which PmCLH1 is a potential chloroplast protein and PmCLH2 may exist as a cytoplasmic protein (Table 2). Even though there may be differences in distributions in cells, their enzymatic activities show that they effectively act on Chls and have a higher preference towards Chl b in vitro (Fig. 4). The substrate preference of full-length PmCLHs was the same as Chlase isolated from an alga (Phaeodactylum tricornutum) (Khalyfa et al. 1995); however, N-terminal-truncated PmCLHs exhibited functional inactivity unlike the in vitro activity of Chlase cloned from citrus (Citrus sinensis) (Harpaz-Saad et al. 2007; Azoulay-Shemer et al. 2008). Accordingly, it is thought that the N-terminal region of PmCLHs contains an important domain for their localization and activity. Furthermore, surfactants, such as Triton X-100, are commonly used in Chlase activity assays to facilitate protein solubilization. The influence of different concentrations of Triton X-100 on the relative Chlase activity was widely investigated (Moll and Stegwee 1978; Yang et al. 2007). According to our enzymatic assay, both PmCLHs efficiently degraded Chls at a final concentration of Triton X-100 of 0.24 %. When Triton X-100 was removed from the reaction buffer, the activities of PmCLHs remained stable but showed a marked change in the substrate preference, especially PmCLH2 (Fig. 4). It is likely that both surfactant-Chl and surfactant-Chlase interactions influence the substrate preference of PmCLHs (Michalski et al. 1987; Fiedor et al. 1992).
It is known that Chlase contains a lipase motif and plays a role as a serine hydrolase (Tsuchiya et al. 2003). However, the crystal structure or three-dimensional structural model analysis of Chlase has never been carried out. Herein, we constructed the predicted tertiary structures of PmCLHs and AtCLHs using the I-TASSER program. The data revealed that PmCLHs and AtCLHs shared a highly similar folding topology, eight β-strands (nine in PmCLH2) flanked by 6–8 α-helices, and the catalytic triad located at the C-terminal end of the central β-strand (Fig. 1). The folding structure adapted to a specific α/β hydrolase fold commonly exists in hydrolytic enzymes (Ollis et al. 1992). The α/β hydrolase fold also forms the core structure of several lipases (Kim et al. 1997). Structural analysis of lipase indicated that a surface loop, named the lid-domain, controls accessibility of the substrate to the catalytic site (Miled et al. 2000). Surprisingly, a similar surface loop structure was found in our modeling, including residues 255–279 in PmCLH1, 240–264 in PmCLH2, 244–271 in AtCLH1, and 245–269 in AtCLH2 (Fig. 1a, b). Thus, we propose that the lid-like structure located downstream of the conserved catalytic histidine residue is important for controlling substrate access to the active site of Chlase.
Abbreviations
- Chlase:
-
Chlorophyllase
- Chl:
-
Chlorophyll
- PPH:
-
Pheophytinase
- Chlide:
-
Chlorophyllide
- Pheide:
-
Pheophorbide
- RT-qPCR:
-
Real-time reverse-transcription quantitative polymerase chain reaction
References
Arkus KAJ, Cahoon EB, Jez JM (2005) Mechanistic analysis of wheat chlorophyllase. Arch Biochem Biophys 438:146–155
Azoulay-Shemer T, Harpaz-Saad S, Belausov E, Lovat N, Krokhin O, Spicer V, Standing KG, Goldschmidt EE, Eyal Y (2008) Citrus chlorophyllase dynamics at ethylene-induced fruit color-break: a study of chlorophyllase expression, posttranslational processing kinetics, and in situ intracellular localization. Plant Physiol 148:108–118
Banas AK, Labuz J, Sztatelman O, Gabrys H, Fiedor L (2011) Expression of enzymes involved in chlorophyll catabolism in arabidopsis is light controlled. Plant Physiol 157:1497–1504
Beneditti CE, Arruda P (2002) Altering the expression of the chlorophyllase gene ATHCOR 1 in transgenic Arabidopsis caused changes in the chlorophyllto- chlorophyllide ratio. Plant Physiol 128:1255–1263
Bruno AK, Wetzel CM (2004) The early light-inducible protein (ELIP) gene is expressed during the chloroplast-to-chromoplast transition in ripening tomato fruit. J Exper Bot 55:2541–2548
Chuang RL, Chen JC, Chu J, Tzen JT (1996) Characterization of seed oil bodies and their surface oleosin isoforms from rice embryos. J Biochem 120:74–81
Distefano G, Las Casas G, Caruso M, Todaro A, Rapisarda P, La Malfa S, Gentile A, Tribulato E (2009) Physiological and molecular analysis of the maturation process in fruits of Clementine Mandarin and one of its late-ripening mutants. J Agr Food Chem 57:7974–7982
Emanuelsson O, Nielsen H, von Heijne G (1999) ChloroP, a neural network-based method for predicting chloroplast transit peptides and their cleavage sites. Protein Sci 8:978–984
Emanuelsson O, Nielsen H, Brunak S, von Heijne G (2000) Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300:1005–1016
Fiedor L, Rosenbach-Belkin V, Scherz A (1992) The stereospecific interaction between chlorophylls and chlorophyllase. Possible implication for chlorophyll biosynthesis and degradation. J Biol Chem 267:22043–22047
Harpaz-Saad S, Azoulay T, Arazi T, Ben-Yaakov E, Mett A, Shiboleth YM, Hortensteiner S, Gidoni D, Gal-On A, Goldschmidt EE, Eyal Y (2007) Chlorophyllase is a rate-limiting enzyme in chlorophyll catabolism and is posttranslationally regulated. Plant Cell 19:1007–1022
Hiba S, Selim K (1997) Biocatalysis of chlorophyllase in ternary micellar systems using pheophytins as substrates. J Chem Technol Biotechnol 68:315–323
Hortensteiner S (2006) Chlorophyll degradation during senescence. Annu Rev Plant Biol 57:55–77
Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K (2007) WoLF PSORT: protein localization predictor. Nucleic Acids Res 35 (Web Server issue):W585-587
Jacob-Wilk D, Holland D, Goldschmidt EE, Riov J, Eyal Y (1999) Chlorophyll breakdown by chlorophyllase: isolation and functional expression of the Chlase1 gene from ethylene-treated Citrus fruit and its regulation during development. Plant J 20:653–661
Kariola T, Brader G, Li J, Palva ET (2005) Chlorophyllase 1, a damage control enzyme, affects the balance between defense pathways in plants. Plant Cell 17:282–294
Khalyfa A, Kermasha S, Marsot P, Goetghebeur M (1995) Purification and characterization of chlorophyllase from alga Phaeodactylum tricornutum by preparative native electrophoresis. Appl Biochem Biotechnol 53:11–27
Kim KK, Song HK, Shin DH, Hwang KY, Suh SW (1997) The crystal structure of a triacylglycerol lipase from Pseudomonas cepacia reveals a highly open conformation in the absence of a bound inhibitor. Structure 5:173–185
Matile P, Schellenberg M, Vicentini F (1997) Localization of chlorophyllase in the chloroplast envelope. Planta 201:96–99
McFeeters RF, Chichester CO, Whitaker JR (1971) Purification and properties of chlorophyllase from Ailanthus altissima (Tree-of-Heaven). Plant Physiol 47:609–618
Michalski TJ, Bradshaw C, Hunt JE, Norris JR, Katz JJ (1987) Triton X-100 reacts with chlorophyll in the presence of chlorophyllase. FEBS Lett 226:72–76
Miled N, Canaan S, Dupuis L, Roussel A, Riviere M, Carriere F, de Caro A, Cambillau C, Verger R (2000) Digestive lipases: from three-dimensional structure to physiology. Biochimie 82:973–986
Moll WAW, Stegwee D (1978) The activity of Triton X-100 soluble chlorophyllase in liposomes. Planta 140:75–80
Nishiyama Y, Kitamura M, Tamura S, Watanabe T (1994) Purification and substrate specificity of chlorophyllase from Chlorella regularis. Chem Lett 23:69–72
Okazawa A, Tango L, Itoh Y, Fukusaki E, Kobayashi A (2006) Characterization and subcellular localization of chlorophyllase from Ginkgo biloba. Z Naturforsch C 61:111–117
Ollis DL, Cheah E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J, Sussman JL, Verschueren KHG, Goldman A (1992) The α/β hydrolase fold. Protein Eng 5:197–211
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Schelbert S, Aubry S, Burla B, Agne B, Kessler F, Krupinska K, Hortensteiner S (2009) Pheophytin pheophorbide hydrolase (Pheophytinase) is involved in chlorophyll breakdown during leaf senescence in Arabidopsis. Plant Cell 21:767–785
Schenk N, Schelbert S, Kanwischer M, Goldschmidt EE, Dormann P, Hortensteiner S (2007) The chlorophyllases AtCLH1 and AtCLH2 are not essential for senescence-related chlorophyll breakdown in Arabidopsis thaliana. FEBS Lett 581:5517–5525
Small I, Peeters N, Legeai F, Lurin C (2004) Predotar: a tool for rapidly screening proteomes for N-terminal targeting sequences. Proteomics 4:1581–1590
Takamiya K, Tsuchiya T, Ohta H (2000) Degradation pathway(s) of chlorophyll: what has gene cloning revealed? Trends Plant Sci 5:426–431
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Terpstra W (1978) Chlorophyllase in Phaeodactylum tricornutum photosynthetic membranes. extractability, small-scale purification and molecular weight determination by SDS-gel-electrophoresis. Physiol Plantarum 44:329–334
Terpstra W (1981) Identification of chlorophyllase as a glycoprotein. FEBS Lett 126:231–235
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl Acids Res 22:4673–4680
Trebitsh T, Goldschmidt EE, Riov J (1993) Ethylene induces de novo synthesis of chlorophyllase, a chlorophyll degrading enzyme, in Citrus fruit peel. Proc Natl Acad Sci USA 90:9441–9445
Tsuchiya T, Ohta H, Masuda T, Mikami B, Kita N, Shioi Y, Takamiya K (1997) Purification and characterization of two isozymes of chlorophyllase from mature leaves of Chenopodium album. Plant and Cell Physiol 38:1026–1031
Tsuchiya T, Ohta H, Okawa K, Iwamatsu A, Shimada H, Masuda T, Takamiya K (1999) Cloning of chlorophyllase, the key enzyme in chlorophyll degradation: finding of a lipase motif and the induction by methyl jasmonate. Proc Natl Acad Sci USA 96:15362–15367
Tsuchiya T, Suzuki T, Yamada T, Shimada H, Masuda T, Ohta H, Takamiya K (2003) Chlorophyllase as a serine hydrolase: identification of a putative catalytic triad. Plant Cell Physiol 44:96–101
Wei Y, Swenson L, Castro C, Derewenda U, Minor W, Arai H, Aoki J, Inoue K, Servin-Gonzalez L, Derewenda ZS (1998) Structure of a microbial homologue of mammalian platelet-activating factor acetylhydrolases: streptomyces exfoliatus lipase at 1.9 Å resolution. Structure 6:511–519
Yamauchi N, Akiyama Y, Kako S, Hashinaga F (1997) Chlorophyll degradation in Wase satsuma mandarin (Citrus unshiu Marc.) fruit with on-tree maturation and ethylene treatment. Sci Hortic 71:35–42
Yang CM, Wang MC, Lu YF, Chang IF, Chou CH (2004) Humic substances affect the activity of chlorophyllase. J Chem Ecol 30:1057–1065
Yang L, Kun A, Xiao Z, Wen-Jun C, Ben-Ke K (2007) AtCLH2, a typical but possibly distinctive Chlorophyllase gene in Arabidopsis. J Integr Plant Biol 49:531–539
Yunyu Y, Selim K, Ronald N (2006) Characterization of sol-gel entrapped chlorophyllase. Biotechnol and Bioeng 95:840–849
Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Chen, M.CM., Yang, JH., Liu, CH. et al. Molecular, structural, and phylogenetic characterization of two chlorophyllase isoforms in Pachira macrocarpa . Plant Syst Evol 300, 633–643 (2014). https://doi.org/10.1007/s00606-013-0908-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00606-013-0908-5