
Abstract
The economically important genus Arachis (Fabaceae) comprises 80 species restricted to South America. One monograph on the genus divided it into nine sections and included an intuitive assessment of evolutionary relationships. There is no comprehensive phylogenetic study of the genus. To test the current systematic treatment of the genus, we reconstructed a phylogeny for Arachis using nuclear ITS and plastid trnT–trnF sequences from 46 species representing all nine sections. ITS cloning of the allotetraploid species of section Arachis indicated the presence of A and B genome alleles and chimeric sequences. Our study revealed that species from section Extranervosae were the first emerging lineage in the genus, followed by sections Triseminatae and Caulorrhizae, and two terminal major lineages, which we refer to as erectoides and arachis. The lineage erectoides comprises members of sections Erectoides, Heteranthae, Procumbentes, Rhizomatosae, and Trierectoides. Species in the arachis lineage form two major clades, arachis I (B and D genomes species and the aneuploids) and arachis II (A genome species). Our results substantiated the sectional treatment of Caulorrhizae, Extranervosae, and Triseminatae, but demonstrated that sections Erectoides, Procumbentes, and Trierectoides are not monophyletic. A detailed study of the genus Arachis with denser taxon sampling, additional genomic regions, plus information from morphology and cytogenetics is needed for comprehensive assessment of its systematics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The genus Arachis (Fabaceae, subfamily Papilionoideae, tribe Dalbergieae) contains approximately 80 species including the economically important cultivated peanut, A. hypogaea (Krapovickas and Gregory 1994; Lavin et al. 2001; Valls and Simpson 2005; Wojciechowski et al. 2004). Krapovickas and Gregory (1994) indicated that the most defining morphological features of the genus are the underground structures, including the fruits, rhizomatous structures, root systems, and hypocotyls. Species of Arachis are distributed east of the Andes Mountains and south of the Amazon River, with the highest diversity around the eastern border of Bolivia and western Brazil (Krapovickas and Gregory 1994). Gregory et al. (1980) cited central Brazil as the center of origin for Arachis and northern Argentina or southern Bolivia as the center of origin of the domesticated peanut. The peanut is among the 30 most important crops that feed the world (Hammer et al. 2003) and is cultivated in more than 100 countries with a 37.1 metric tons global production. Other species of Arachis, for example A. villosulicarpa, have also been cultivated as indigenous food crops (Galgaro et al. 1997). In addition, A. glabrata, A. pintoi, and A. repens are grown as forage crops (Galgaro et al. 1998).
There is no comprehensive molecular systematic study of the genus. The latest monograph on Arachis was based on morphology, geographic distribution, cytogenetics, and hybridization success (Krapovickas and Gregory 1994). It recognized 69 species, dividing them into nine sections: Arachis, Caulorrhizae, Erectoides, Extranervosae, Heteranthae, Procumbentes, Rhizomatosae, Trierectoides, and Triseminatae. Recently, 11 additional species were recognized in Arachis (Valls and Simpson 2005). Although these studies advanced our knowledge on sectional classifications, ambiguities remain in Arachis systematics and phylogenetics. The section Arachis is the largest, containing 31 species, or 45% of all currently recognized species (Krapovickas and Gregory 1994; Valls and Simpson 2005). It is the most studied of the nine sections because it contains the cultivated peanut and its gene pools (Singh and Simpson 1994). The section is further divided into three groups of species based on genome type: A, B, or D (Smartt et al. 1978; Stalker 1991), with the allotetraploids cultivated A. hypogaea and wild A. monticola containing AB genomes. The phylogenetic relationships among these species are not fully understood.
Most of the Arachis species are diploid based on x = 10 (Krapovickas and Gregory 1994). However, there are five tetraploid species (2n = 4x = 40) and four aneuploid species (x = 9, 2n = 2x = 18) (Krapovickas and Gregory 1994; Lavia 1998; Peñaloza and Valls 1997). Two of the tetraploids (A. hypogaea and A. monticola) are in sect. Arachis, and the remaining three are in sect. Rhizomatosae (A. glabrata, A. pseudovillosa, and A. nitida) (Krapovickas and Gregory 1994; Valls and Simpson 2005). Three of the aneuploids, A. decora, A. palustris, and A. praecox, belong to sect. Arachis (Lavia 1998; Peñaloza and Valls 1997), but their genomic identity and phylogenetic position has not been established. The fourth aneuploid, A. porphyrocalyx, is placed in sect. Erectoides (Peñaloza and Valls 2005).
Krapovickas and Gregory (1994) presented an illustration of intuitive “evolutionary and phylogenetic relationships” among the nine sections of Arachis based on information from crossability, morphology, and geography. They concluded that Erectoides, Extranervosae, Heteranthae, Trierectoides, and Triseminatae are “older” sections whereas Arachis, Caulorrhizae, Procumbentes, and Rhizomatosae are more “recent” in origin. They indicated that species from sect. Erectoides produce hybrids with members of other sections. Although most intersectional hybrids are sterile, Krapovickas and Gregory (1994) considered sect. Erectoides to be less isolated than the other sections. Other studies provided cytogenetic evidence of closer genomic relationships between Erectoides and most of the remaining sections (Gregory and Gregory 1979; Stalker 1981; Valls and Simpson 1994). However, Krapovickas and Gregory (1994) maintained that the use of cytogenetic studies to elucidate the phylogenetic relationships among the sections and species is insufficient because of the genetic barriers among Arachis species caused by geographic isolation and an autogamous reproductive system.
Relationships among species and sections of Arachis have been evaluated using molecular markers such as restriction fragment length polymorphism (RFLP) and random amplification of polymorphic DNAs (RAPD) (Barkley et al. 2007; Creste et al. 2005; Galgaro et al. 1998, 1997; Gimenes et al. 2002b). All of these studies were based on phenetic analyses using the unweighted pair group method using arithmetic averages (UPGMA). Some of these studies supported the Krapovickas and Gregory (1994) classification whereas others raised questions concerning the validity of some sections, for example sect. Heteranthae and Rhizomatosae. Hoshino et al (2006) were the first to include all nine sections of Arachis in a phenetic analysis (UPGMA) of microsatellite data. Most of the species were grouped in their respective sections as defined by Krapovickas and Gregory (1994). However, some of sect. Procumbentes species grouped with species from sect. Erectoides, and a few grouped with sections Trierectoides and Heteranthae. Their study also showed that members of sect. Heteranthae failed to group together, which is in agreement with the findings of Galgaro et al. (1998).
Two studies used DNA sequence information to examine species relationships in Arachis, but the focus was on sect. Arachis and the putative progenitors of A. hypogaea (Jung et al. 2003; Tallury et al. 2005). Jung et al. (2003) used sequences of stearoyl-ACP and oleoyl-PC desaturases from seven species of sect. Arachis and analyzed the data using maximum parsimony (MP). Their unrooted tree provided further support for the origin of the A and B genomes of A. hypogaea from A. duranensis and A. ipaensis, respectively. Tallury et al. (2005) used plastid trnT–trnF sequences to examine the evolutionary relationships among the genomes in sect. Arachis and the affinities of the aneuploid species for these genomes. Using maximum likelihood (ML), the unrooted tree showed that the B and D genome species are more closely related to each other than to the A genome species, and that the aneuploids are closer to the B and D genome species.
We present here a molecular phylogenetic study representing all nine sections and recognized genomes of the genus Arachis using sequence information from two intergenic spacers, one intron of the plastid trnT–trnF region, and both nuclear ribosomal internal transcribed spacers (ITS1 & 2). DNA sequence information from these regions has been successfully used to depict the phylogenetic relationships among species within genera of Fabaceae (Tun and Yamaguchi 2007; Vander Stappen et al. 2002). The data were analyzed phylogenetically using maximum parsimony and Bayesian inference. We also applied TCS network analysis to assess relationships among genomes and alleles within the ITS region, because of the presence of diverse sets of haplotypes, including potential chimeras.
Materials and methods
Taxon sampling
We analyzed a total of 48 accessions representing 46 species from all nine sections of Arachis recognized by Krapovickas and Gregory (1994). Stylosanthes humilis and S. fruticosa were chosen as outgroups on the basis of their sister group relationship to Arachis (Lavin et al. 2001). We generated new ITS and trnT–trnF sequences for 34 accessions and supplemented the ITS datasets with 14 GenBank sequences. These GenBank sequences did not have corresponding trnT–trnF sequences and genomic DNA was not available for them. Consequently, we generated two datasets for the ITS, one that included the 34 accessions plus four clones sequenced in our laboratory, which matches exactly the trnT–trnF dataset, and the second included all 48 accession plus the four clones, which we will refer to as the “extended dataset”. Species examined, sectional affiliation, and sources of material are listed in Table 1.
DNA extraction, amplification, cloning, and sequencing
Genomic DNA extraction followed Milla et al. (2005) using fresh leaf material. The ITS region, including ITS1, 5.8S, and ITS2, was amplified using primers ITS4 and ITS5 (White et al. 1990) in a 25-μl reaction mixture containing 1× ThremoPol buffer (New England Biolabs, Ipswich, MA, USA), 200 μM dNTPs, 20 pmol of each primer, and 1.5 U Taq DNA polymerase. The amplifications were carried out in a PTC-100 (MJ Research, Waltham, MA, USA) with a 2-min initial denaturing step at 94°C, 35 cycles of 2 min denaturing at 94°C, 1 min of primer annealing at 52°C, and 1.5 min extension at 72°C, followed by a final extension of 5 min at 72°C.
Amplifications of the trnT–trnF region were carried out using universal primers (Taberlet et al. 1991) as described in Tallury et al. (2005). Primers rps4-166F and trnL-P6/7 (Quandt et al. 2004) were used to generate amplified products when the universal primers failed to amply the trnT–trnL region. PCR-amplified products for the ITS and trnT–trnF regions were resolved on 1% TAE-agarose gel containing 0.03 mg ethidium bromide and were cleaned using QIAquick PCR purification or QIAquick gel extraction kits (Qiagen, Valencia, CA, USA). Cycle-sequencing reactions were carried out using ABI Big Dye Terminator Ready Reaction (Applied Biosystems, Foster City, CA, USA), following standard procedures, and then electrophoresed on an Applied Biosystems 3730 automated sequencer at the Core Sequencing Facility at VBI, Virginia Tech, VA, USA, or at the DNA Analysis Facility of Duke University, NC, USA.
The amplified ITS regions from the allotetraploid species A. hypogaea and A. monticola were ligated into the pGEM-T Easy vector (Promega, Madison, WI, USA) to separate multiple alleles suspected from double peaks in pherograms from direct sequencing of PCR amplifications of genomic DNAs. Ligated plasmids were heat shocked into JM109 Escherichia coli cells (Promega). Positive colonies were detected via blue–white screening and grown using standard procedures. Positive clones were grown and DNA was isolated by the plasmid miniprep procedure (Birnboim and Dolye 1979). Inserts were sequenced directly using the vector specific primers. When direct sequencing failed, the ITS insert was amplified from the clones using the ITS primers, and the amplified inserts were cleaned and sequenced.
Sequence alignment and phylogenetic analysis
Sequences were manually aligned using PhyDE (Müller et al. 2005) following the alignment rules in Kelchner (2000). One identified inversion in the trnT–trnL spacer was positionally separated in the alignments, and later included as reverse complement in the phylogenetic analyses, as discussed by Quandt et al. (2003) and Borsch and Quandt (2009). Indels were coded using SeqState (Müller 2005) following the simple indel coding method (Simmons and Ochoterena 2000). The datasets were analyzed with indels coded (IC) as binary characters and without the use of indels (IN). Aligned datasets for the nuclear ITS (excluding the 26S exon) and plastid trnT–trnF were analyzed separately and in combination. Because the plastid genome in Arachis is maternally inherited (Tallury et al. 2005), only the maternal ITS region as identified by the TCS analysis was used for the allotetraploid species in the combined analysis. Phylogenetic analysis of the three datasets was conducted using maximum parsimony in PAUP* (Swofford 2003) and Bayesian inference (BI) in MrBayes (Huelsenbeck and Ronquist 2001). Maximum parsimony analyses were performed as heuristic tree searches using tree bisection–reconnection (TBR) branch-swapping with 1,000 random addition sequence replicates, and incorporating the parsimony ratchet algorithm (Nixon 1999) via PRAP2 (Müller 2007). A strict consensus tree was generated for each dataset. Support for the clades was obtained by performing bootstrap (BS; Felsenstein 1985) searches with 1,000 replicates and 10 random sequence replicates. Bayesian analysis was performed using the GTR + Γ+ I models, with 1,000,000 generations and 10 runs. Trees were compiled using TreeGraph2 (Müller and Müller 2004; Stöver and Müller 2010).
A partition homogeneity test (PHT; Farris et al. 1995) was performed in PAUP* version 4.0b10 (Swofford 2003) before combined analyses to determine if the ITS and trnT–trnF regions have significantly different phylogenetic signals. The PHT was conducted using 100 replicate partitions. For each replicate partition, no more than 500 trees were saved with a score greater than 1. The resulting P value (0.01) indicated that the two regions are incongruent, which implied that the ITS and trnT–trnF regions should not be combined. However, PHT has been found to be sensitive to noise and different mutation rates between genomic regions, and might not be an accurate measure of congruence between regions (Baker and Lutzoni 2002; Darlu and Lecointre 2002; Dolphin et al. 2000). To assess the potential sources of conflict, the matrices for the two genomic regions were analyzed separately and in combination. Comparison of trees derived from the partitioned analyses showed that support for conflicting nodes was low, implying soft incongruence (Johnson and Soltis 1998).
Haplotype networks were constructed using TCS (Clement et al. 2000, 2002). Because only singleton gaps were observed in the data, the default settings were applied. The TCS analysis enabled identification of chimeric ITS sequences that were subsequently excluded from the phylogenetic analyses.
Results
ITS alleles and sequence statistics
Amplification and sequencing of the ITS region generated a single homogenous sequence in each accession with the exception of the two allotetraploid species A. hypogaea and A. monticola. In these allotetraploids, the chromatograms depicted a prominent signal for the putative A genome allele and secondary peaks appearing as possible background. Consequently, the genomic ITS PCR products of the tetraploids were cloned and sequenced. Fourteen clones were obtained from each of A. hypogaea and A. monticola. For A. hypogaea, most of the clones corresponded to the ITS sequence of the A genome species (10 clones; 71.4%) whereas two clones matched the B genome species (14.3%; remaining two clones were chimeric as noted below). For A. monticola, six clones (42.9%) represented the A genome whereas two clones (14.3%) represented the B genome (remaining six clones were chimeric as noted below). Most of the A genome clones clustered with the A. duranensis haplotype, whereas those with the B genome grouped with the A. ipaensis haplotype in the TCS analysis (Supplementary Fig. 1). There were a few clones in which the haplotype differed from either A. duranensis or A. ipaensis because of mutations in a few positions. When the alignment of the A and B genome clones was examined, the positions where mutational differences were observed corresponded exactly to the sites of the double peaks. The two alleles differed by eight substitutions and a single-base indel. The lower frequency of the B allele in the allotetraploids may have caused the lower signal (smaller peaks) in the chromatograms, whereas the one-base gap resulted in a chromatogram shift that resembled background noise.
Alleles with chimeric sequences (total of seven clones, 24%) were also observed in both allotetraploids. These included three types of chimera that differ in the genomic identity of ITS1 and ITS2 regions:
-
1
ITS1 represents the A allele whereas ITS2 represents the B allele (two clones, 14.3%);
-
2
ITS1 represents the B allele whereas ITS2 represents the A allele (one clone, 7.1%); and
-
3
one of the ITS regions is a hybrid of the A and B genomes whereas the other represents one of these two genomes (five clones; 35.7%).
The complete ITS region sequences varied in length from 599 bp in A. brevipetiolata to 657 bp in A. hypogaea. The sequences were trimmed at the 26S rDNA subunit, 63 bp, as these sequences were incomplete for this exon. Twelve indels of 1–9 bp were required for the alignment of all the species including the outgroup taxa, resulting in a total alignment of 614 characters. Of these characters, 153 (25%) were variable, of which 97 (63.4%) were parsimony informative (PI). The inclusion of indels in the phylogenetic analyses increased the number of total characters to 634, the number of variable characters to 174 (27.4%), and the PI characters to 108 (62.1%).
ITS-based phylogeny
The MP analysis of the 34-taxon dataset that included the sequences of the cloned alleles of the allotetraploid species resulted in seven most parsimonious trees of 221 steps (indels excluded) and six trees of 196 steps (indels included). The consistency index (CI) was 0.913 and the retention index (RI) 0.938 without indels and 0.905 and 0.932 with indel coding (Table 2). The consensus trees derived from the analyses of these two datasets were identical in topology but differed in BS support (Fig. 1). The Bayesian analyses with and without indels resulted in 50% majority trees that are completely congruent in topology and were identical to the MP consensus trees, differing in degrees of support for some nodes (Fig. 1). Because topology was the same for MP and BI trees, we present the BI tree with both BS and posterior probability (PP) support noted on the branches (Fig. 1).

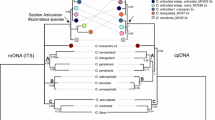
Bayesian inference 50% phylogeny majority rule consensus trees for Arachis rooted with Stylosanthes humilis and S. fruticosa and using the GTR + Γ+ I model based on sequences from the nuclear ribosomal ITS region (left) and the trnT–trnF region (right). Arrows indicate branches that collapsed when the IUPAC characters were included for the allotetraploid ITS sequences in the dataset. Triangles indicate branches were not recovered in MP analyses. Plus symbols indicate branches not recovered in MP analyses with indel coding. Filled stars indicate branches not recovered in MP analyses without indel coding. Filled diamonds indicate branches not recovered in the BI analysis when indel characters were included. Posterior probabilities are above the branches and bootstrap support values are below. Supports with indels coded are on the left and those obtained without indel coding are on the right
The monophyly of the genus received maximum PP and BS support in all cases (Fig. 1). Arachis macedoi (sect. Extranervosae, Ex genome) was sister to the remaining Arachis (0.95 PP-IN, 0.89 PP-IC, 62% BS-IN, 54% BS-IC) followed by A. triseminata (sect. Triseminatae, T genome), excluded by maximum PP support and 99% BS-IN and 100% BS-IC support. A clade comprised of members of sect. Caulorrhizae (C genome), A. pintoi plus A. repens, diverged next (1.00 PP-IN and PP-IC; 95% BS-IN, 99% BS-IC). Exclusion of section Caulorrhizae from the other Arachis received moderate to low BS support (76% IN, 68% IC) but maximum PP support. The next lineage (1.00 PP-IN and PP-IC, 85% BS-IN, 93% BS-IC) comprises species placed by Krapovickas and Gregory (1994) into sections Erectoides, Heteranthae, Procumbentes, and Trierectoides. We will refer to this clade as group erectoides. Components of this clade split into two subclades. One comprised a polytomy of A. guaranitica and A. tuberosa (sect. Trierectoides) plus A. major 559 (sect. Erectoides) (0.93 PP-IN, 0.90 PP-IC, <50% BS). The other subclade of group erectoides (0.97 PP-IN, 0.96 PP-IC, 77% BS-IN, 79% BS-IC) was indicative of a polytomy between A. paraguariensis (sect. Erectoides), A. appressipila (sect. Procumbentes), and a clade of A. dardani (sect. Heteranthae) + A. rigonii (sect. Procumbentes) (1.00 PP-IN and PP-IC, 87% BS-IN and BS-IC).
Following erectoides, the remaining species of the genus formed two terminal clades, which we hereafter will refer to as groups arachis I and arachis II (Fig. 1). The sister relationship of these two groups received moderate BS support (68% BS-IN, 71% BS-IC), but PP values were significant, i.e., ≥ 0.95 (0.97 PP-IN, 0.96 PP-IC). Group arachis I was strongly supported (1.00 PP-IN and PP-IC, 98% BS-IN and BS-IC) and included sect. Arachis aneuploid species, B and D genome species, and B genome clones of the allotetraploids. The aneuploid species A. praecox and A. palustris formed a fully supported subclade sister to the other species, excluded by 86% BS-IN, 87% BS-IC, 0.99 PP-IN and PP-IC. Among the remaining arachis I species, the B genome species A. ipaensis, A. magna, A. williamsii, A. valida, and the B genome clones for the allotetraploid species appeared in a polytomy in a weakly supported clade (0.97 PP-IN and PP-IC, 65%BS-IN, 63% BS-IC). Another subclade showed the D genome species A. glandulifera in a weakly supported sister relationship to a fully supported subclade that included the B genome species A. cruziana and A. batizocoi. The A genome species A. trinitensis and A. benensis formed a polytomy with the two above described subclades of arachis I. The second terminal clade, arachis II, comprised the remaining A genome species of sect. Arachis, although BS support was low and PP values were not significant (0.65 PP-IN and 0.86 PP-IC, 66% BS-IN and 67% BS-IC). At the base of the clade (Fig. 1), A. diogoi and A. hoehnei (sect. Arachis) and A. major 10588 (sect. Erectoides) appeared in a polytomy sister to a subclade (0.92 PP-IN, <50% BS) that included sect. Arachis “A” genome species. This clade included the A genome clones for the allotetraploids, A. hypogaea and A. monticola along with putative A genome donor A. duranensis.
The alignment of the expanded dataset was 619 characters in length not including indels. Among these characters, 171 (27.6%) are variable and 117 (68.4%) are PI. Two additional indels were identified in the ingroup taxa in the extended dataset. When indels were included, the number of variable characters increased to 204 (31.3%) and the PI characters to 133 (65.2%). The MP analysis without indel coding identified 15 trees of 238 steps. The CI and RI for this analysis were 0.861 and 0.929, respectively. When indels were scored as characters, the MP analyses resulted in 58 most parsimonious trees of 277 steps, and CI and RI values of 0.859 and 0.925, respectively. The overall topologies of the consensus trees were the same with and without the inclusion of indels, differing in a slight increase in resolution within the arachis II clade when indels were included and in arachis I when indels were excluded (Fig. 2). The monophyly of the genus received maximum support in all analyses. Members of sect. Extranervosae (A. burchellii, A. macedoi, A. lutescens, and A. villosulicarpa) formed a clade with maximum PP and BS support that appeared sister to remaining sections, excluded by 1.00 PP-IN, 0.99 PP-IC, 79% BS-IN and 60% BS-IC. The next diverging lineage was the monotypic sect. Triseminatae, excluded from the remaining species by 99% BS-IN and BS-IC support and maximum PP. Section Caulorrhizae (93% BS-IN, 99% BS-IC, maximum PP) diverged next, followed by A. burkartii, the only known diploid species from sect. Rhizomatosae (Fig. 2). Support for the exclusion of A. burkartii from the two remaining lineages was low. The remaining clades, erectoides and arachis I plus arachis II appeared next. Topology of these latter clades was congruent with that obtained with the ITS dataset.

Arachis phylogeny generated using Bayesian inference based on ITS expanded dataset, indels coded as characters, and the GTR + Γ+ I model. Phylogenies generated using MP were nearly identical with the Bayesian tree in topology. Plus symbols indicate branches not recovered in MP analyses with indel coding. Filled stars indicate branches not recovered in MP analyses without indel coding. Filled diamonds indicate branches not recovered in the BI analysis when indel characters were included. Posterior probabilities are above the branches and bootstrap support values are below. Supports with indels coded are on the left and those obtained without indel coding are on the right
trnT–trnF based phylogeny
The complete sequences of the trnT–trnF region varied in length from 1,789 to 1,960 bp. Seventeen indels of 1–22 bp were required for the alignment of all the species including the outgroup taxa, resulting in a total alignment of 2,052 characters. Excluded were 117 characters that included the 5′ trnL, 23 characters from the adjacent trnT–L spacer, 34 from the trnL intron, and three small homopolymeric repeats and one tandem repeat region because of a large number of missing data and alignment/coding ambiguities. Of the remaining 1,935 characters, 202 (10.4%) were variable, of which 114 (56.4%) were parsimony informative. The inclusion of indels in the phylogenetic analyses increased the number of total characters to 1995, the number of variable characters to 261 (13.1%), and the PI characters to 152 (58.2%).
MP analysis resulted in 31 most parsimonious trees of 241 steps from the non-indel coded dataset and 391 trees of 336 steps from the indel coded dataset. The CI values were 0.892 without indels and 0.815 with indels, and the RI values were 0.912 with and 0.854 without indels. The consensus trees constructed from these 31 trees were identical in topology with that derived from the 336 trees, differing in BS support (Fig. 1). The Bayesian analyses with and without indels resulted in phylogenetic trees that are completely congruent in topology with each other and with the MP consensus trees, but differed in degrees of support for the nodes. Consequently, the tree obtained from the Bayesian analysis with PP and BS support will be presented.
As in the ITS analyses, the monophyly of the genus received full PP and BS support in the trees based on the trnT–trnF data. A basal clade emerged comprising Triseminatae sister to Extranervosae plus Caulorrhizae; however, support for this clade and its internal structure was low (Fig. 1). This basal clade was sister to a strongly supported terminal clade that included arachis I and arachis II plus erectoides; support for the sister group relationship of arachis II and erectoides was not significant. The aneuploid species did not appear monophyletic (Fig. 1). The arachis I species fell into two moderately supported major subclades.
The arachis II clade received high BS support (98% BS-IN, 99% BS-IC) and maximum PP support. New subclades emerged with the trnT–trnF data, for example the A-genome species A. diogoi, A. hoehnei, and A. correntina (1.00 PP-IN, 0.83 PP-IC, 64% BS-IN, <50% BS-IC) and a subclade comprising A. kempff-mercadoi, A. helodes, and A. herzogii (0.66 PP-IN, <50% BS-IN and BS-IC). Remaining arachis II species formed a polytomy with these subclades (Fig. 1).
Combined ITS and trnT–trnF analysis
Analyses of the combined ITS and trnT–trnF sequences resulted in a total alignment of 2,547 characters, of which 355 (13.9%) were variable and 211 (59.4%) parsimony informative. With the inclusion of indels, the total number of characters increased to 2,628; of these, the number of variable characters was 435 (16.6%) and the PI characters were 260 (60.0%). MP analysis of the combined dataset without indels recovered 167 best trees of 446 steps, whereas with inclusion of the indels in the coded dataset 540 best trees of 568 steps were obtained. The CI and RI were relatively high, but decreased slightly with inclusion of the indels (Table 2). Strict consensus trees constructed from both of these MP analyses were identical in topology with each other and with the BI tree (trees not shown).
The monophyly of the genus Arachis was fully supported. The backbone topology of the tree based on the combined dataset was identical with that obtained with the extended ITS dataset, differing in resolution within groups arachis I and erectoides (Fig. 1). The structure of the erectoides clade was similar to that resolved with the trnT–trnF data whereas the relationships within groups arachis I and arachis II were similar to those in the ITS-based tree. Support was strong to maximum for the monophyly of these three terminal clades.
Discussion
Molecular evolution
Generally, it is believed that the ITS region concerts towards one of the parental progenitors in hybrids and allopolyploids because it undergoes rapid homogenization (Álverez and Wendel 2003; Soltis et al. 2008). Considering the signal in the pherograms obtained from the direct genomic sequencing of the allotetraploid species A. hypogaea and A. monticola, the A genome ITS allele seemed to be the more dominant in positions where double peaks representing the two alleles were found. This may be explained by the presence of the A allele clones at much higher frequency than the B allele clones (71.4 vs. 14.3% for A. hypogaea and 42.9 vs. 14.3% for A. monticola), implying that the ongoing process of concerted evolution favors the A allele over the B allele. Although the occurrence of multiple alleles has been frequently documented (Kovarik et al. 2004; Soltis et al. 2008; Vander Stappen et al. 1998), their detection can sometimes go unnoticed, because the region contains few polymorphic sites indicative of each genome (Soltis et al. 2008), or a low signal for one of them, as shown in this study. This study has also shown the presence of chimeric alleles in six of the 14 (42.8%) A. monticola clones and in two of the 14 A. hypogaea clones (14%), implying the occurrence of infra-allelic recombination events in the ITS of both tetraploid genomes. The presence of chimeric alleles was confirmed in our TCS analysis of the ITS1 and ITS2 alleles from diploid species in sect. Arachis and the cloned alleles from the tetraploid species (Supplementary Fig. 1). These chimeric alleles were closely linked with the respective ITS regions of their parental alleles, as demonstrated by the TCS haplotype network (Supplementary Fig. 1). The evolutionary dynamics in the Arachis ITS region as illustrated by the chimeric ITS may represent a fluid state that will subsequently undergo fixation by concerted evolution towards one of the ancestral genotypes.
Without cloning, nucleotide sites showing multiple peaks in the direct genomic sequences of taxa with a multiallelic ITS region are usually assigned IUPAC ambiguity codes. Soltis et al. (2008) have demonstrated that phylogenetic reconstruction will be negatively affected when IUPAC ambiguity codes are used as molecular characters. This point received further support from our study. Inclusion of the nine IUPAC coded characters for the allotetraploid species in a dataset that has a total of 153 variable characters and 97 PI characters resulted in substantial collapse of nodes in the clades containing the tetraploids (arachis I and arachis II clades; see arrows denoting collapsed nodes in Fig. 1). However, when cloned sequences representing the A and B alleles were used alone to represent the tetraploid species in the MP analysis, resolution improved substantially and the clones grouped with the sequences of the respective putative diploid progenitors (Fig. 1). The collapse of nodes illustrates the effect of using ambiguous sequence characters caused by unrecognized multiple copies in the genome, and stresses how carefully ITS data should be treated in general. When ambiguous sites are detected, cloning of this region is highly recommended.
Phylogeny of Arachis
The Arachis phylogenetic trees based on analyses of partitioned ITS and trnT–trnF sequence data produced two alternative topologies (Fig. 1). The major differences resided in the backbone of the trees, namely the identity of the first emerging lineages and the position of the Erectoides group. With the ITS data, A. macedoi (sect. Extranervosae) diverged first followed by A. triseminata and then sect. Caulorrhizae (A. pintoi and A. repens; Fig. 1). In the trnT–trnF tree, the same three lineages appeared at the base in a single clade, although support was low, rendering the basal topology here rather ambiguous. Regarding the placement of erectoides, its sister group relationship with arachis II with the trnT–trnF region received <50% BS and non-significant PP support. This topological difference is attributed to soft incongruence. The phylogenetic tree based on combined datasets resulted in full resolution for all major clades, including the basal ones, and strong to moderate support for other clades (tree not shown). Analyses of the extended ITS dataset recovered phylogenies that were topologically congruent with those obtained with the combined dataset (Fig. 2). Therefore, for the discussion of the phylogenetic relationship and subsequent systematic implication, we will use the tree based on the extended ITS dataset. However, species relationships will be discussed in the light of their placement based on the maternally inherited trnT–trnF and bipaternally inherited ITS regions.
Basal grade
The tree based on the extended ITS dataset resolved species of sect. Extranervosae (A. burchellii, A. lutescens, A. macedoi, and A. villosulicarpa) at the base followed by a grade of sect. Triseminatae and sect. Caulorrhizae (Fig. 2). Gimenes et al. (2002b) showed A. burchelli and A. macedoi in a cluster with 81% BS support in a UPGMA analysis of RFLP data, providing support for the monophyly of sect. Extranervosae. The phylogenetic position for sect. Extranervosae is in disagreement with the phylogenetic notions of Krapovickas and Gregory (1994). They indicated that although sect. Extranervosae is a “presumably older section”, sect. Trierectoides is “probably the most primitive [section] of the genus”. They based their conclusion on the assumption that sect. Trierectoides is “genetically very isolated” from the remaining sections of the genus. In this study, the two species of sect. Trierectoides were nested deeper in the tree within the group erectoides (Fig. 2).
The position of the second diverging lineage, sect. Triseminatae, was strongly supported in all analyses, including the trnT–trnF tree where it appeared in a clade with two other basal sections (Figs. 1, 2). Krapovickas and Gregory (1994) considered this section to be one of the “older” sections in the genus because it is genetically isolated. Their assessment was based on unsuccessful attempts to generate hybrids in crosses between sect. Triseminatae and species from other sections. The UPGMA analysis of microsatellite markers resolved accessions of A. triseminata into a cluster distinct from the remaining species of the genus (Hoshino et al. 2006). The affinity between sections Triseminatae and Caulorrhizae has previously been demonstrated by UPGMA analysis of RAPD and RFLP data (Galgaro et al. 1998). The placement of the third diverging lineage, sect. Caulorrhizae (A. pintoi and A. repens) was evident in all analyses (Figs. 1, 2). The monophyly of the section received support from studies based on hybridization and pollen fertility, microsatellite data, RFLP, and isozymes (Gimenes et al. 2002b; Gregory and Gregory 1979; Hoshino et al. 2006; Palmieri et al. 2005). Species of this section are easily distinguishable, on the basis of leaf shape and leaflet size (Gimenes et al. 2002b; Krapovickas and Gregory 1994), from other species of the genus.
Erectoides
Group erectoides has not been previously recovered as such (Figs. 1, 2). This is a heterogeneous lineage containing representatives of sections Erectoides, Heteranthae, Procumbentes, Rhizomatosae, and Trierectoides, although none of the sections appeared monophyletic. UPGMA analysis of AFLP data (Gimenes et al. 2002b) recovered a similar group composed of species from sections Erectoides, Procumbentes, Heteranthae, and the tetraploid A. glabrata of sect. Rhizomatosae. Using microsatellite data, Hoshino et al. (2006) obtained a cluster of species from sections Erectoides and Procumbentes only. The emergence of erectoides in this study can be explained in a historic perspective. Section Trierectoides was previously treated as part of sect. Erectoides, but was later segregated out, on the basis of the presence of three leaflets instead of the four leaflets found in all other sections (Krapovickas and Gregory 1994). Species currently recognized as sect. Procumbentes were also historically included in sect. Erectoides, but were removed by Krapovickas and Gregory (1994) on the basis of decumbent habit and the development of flowers along the branches. Therefore, except for sect. Heteranthae, it is not surprising to see the convergence of these taxa into one lineage with strong support.
Arachis I
Members of sect. Arachis appeared in one cluster in phenetic analyses (Gimenes et al. 2002b; Milla et al. 2005), or a well-supported lineage in phylogenetic analyses (Tallury et al. 2005). The phylogenetic structure of Arachis I is comparable with that recovered by Tallury et al. (2005), demonstrating the sistergroup relationship between the B and D genome species (0.99 PP-IN, 1.00 PP-IC, 86% BS-IN, 87% BS-IC) with the aneuploids being sister to both. The phylogenetic placement of the aneuploids, which received maximum to strong support, has not been previously resolved (Figs. 1, 2).
B genome
Species of sect. Arachis with the B genome formed two well supported subclades in arachis I based on trnT–trnF and combined sequence data with high to maximum PP support and good to high BS support (Fig. 1). However, these two clades did not emerge in the analyses of the ITS datasets (Figs. 1, 2). This difference in structure may reflect the pattern of inheritance of these two genomic regions. The B genome is identified by the lack of the small “A” chromosome characteristic of the A genome (Fernández and Krapovickas 1994; Krapovickas and Gregory 1994). The emergence of A. ipaensis and A. batizocoi with A. cruziana in one subclade based on combined data analyses was unexpected. Arachis ipaensis has previously been regarded as more closely related to A. magna than to either A. batizocoi or A. cruziana on the basis of data from crossability (Burow et al. 2009), RAPD and RFLP (Gimenes et al. 2002a), AFLP (Milla et al. 2005), the sequence of nuclear hypervariable regions (Moretzsohn et al. 2004), and microsatellite markers (Bravo et al. 2006). The grouping of A. batizocoi and A. cruziana in this study (Figs. 1, 2) received support from studies based on AFLP (Milla et al. 2005), RFLP and cytogenetic data (Burow et al. 2009), and plastid sequence data (Tallury et al. 2005). Placement of A. ipaensis with A. cruziana and A. batizocoi was also demonstrated previously in the ML network based on trnT–trnF sequence data, although BS support was weak (Tallury et al. 2005).
D genome
Only A. glandulifera has been designated to have the D genome (Stalker 1991). The placement of this species in arachis I received mostly strong support in analyses of partitioned and combined datasets (Figs. 1, 2). However, its position within the clade differed, depending on the genomic region used. The ITS data showed low support for its placement as sister to A. batizocoi and A. cruziana, whereas the trnT–trnF data included it in the subclade containing A. magna, A. williamsii, A. benensis, A. trinitensis, and A. valida with moderate BS and full PP support (Fig. 1). Tallury et al. (2005) resolved a moderately supported lineage that included A. glandulifera with A. williamsii and A. benensis; they did not include A. magna, A. valida, and A. trinitensis in their study. Therefore, although a common ancestry between the B and D genome species is evident, the exact relationship between them remains unclear.
Section Arachis aneuploids
The original placement of the three aneuploid species A. decora, A. praecox, and A. palustris (2n = 2x = 18) in sect. Arachis was based on morphology and geographical distribution (Krapovickas and Gregory 1994). Only a few studies focusing on sect. Arachis have included these aneuploid species (Bravo et al. 2006; Creste et al. 2005; Gimenes et al. 2007; Milla et al. 2005; Tallury et al. 2005). Microsatellite data placed accessions of these species together in a cluster with the A genome species A. diogoi, A. kuhlmannii, and A. simpsonii (Bravo et al., 2006). In AFLP based dendrograms, the aneuploids formed a separate cluster (Milla et al. 2005; Tallury et al. 2005). However, in the trnT–trnF study of Tallury et al. (2005), and in this study, the aneuploid species appeared sister to the B and D genome clade and were excluded from the A genome species with strong BS and PP support, implying higher genetic affinities to the B and D genomes than the A genome (Figs. 1, 2). The phylogenetic placement of the aneuploids points to chromosomal evolution following the split of arachis I and arachis II from a common ancestor (Figs. 1, 2). This event resulted in two sister lineages representing the aneuploids (2n = 18) and the mostly B genome species that maintained the common chromosome complement of 2n = 20. This interpretation is more parsimonious than loss of a pair of chromosomes in the aneuploids and their being restored in the mostly B genome lineage.
Arachis II
This group contains all A genome species of sect. Arachis plus A. major 10588 (sect. Erectoides) (Figs. 1, 2). Lack of resolution is evident in the arachis II clade in this study and in previous studies (Gimenes et al. 2002b; Tallury et al. 2005). The low resolution and support among members of arachis II may imply recent origin or low rate of substitution. The placement of an accession of A. major (sect. Erectoides) within arachis II was not unexpected. Krapovickas and Gregory (1994) had noted that crosses among A. major accessions from the farthest points of its distribution did not produce fertile hybrids. Thus, they suggested that although the accessions seem to be morphologically similar, it was possible that they belong to different biological species. Therefore, A. major is in need of a detailed study at the population level to determine species boundaries and its phylogenetic placement in the genus.
Origin of the allotetraploids in sect. Arachis
The different inheritance of the ITS and trnT–trnF regions provided insight into the evolutionary history of the allotetraploids A. hypogaea and A. monticola. The trnT–trnF analysis confirmed the maternal origin of the A genome, because the allotetraploids appeared as part of the A genome lineage but in a polytomy (Figs. 1, 2). The ITS clones further substantiated the results of the trnT–trnF data and provided evidence for the paternal origin of the B genome (Figs. 1, 2) as previously noted by Kochert et al. (1991), Jung et al. (2003), and Seijo et al. (2004). Although the presence of polytomies prevented us from pinpointing the exact species donors, the putative donor species, A. duranensis and A. ipaensis, were recovered in those polytomies (Figs. 1, 2). Therefore, better resolution and support using more genomic regions with higher substitution is needed before molecular phylogenetics can provide support for the ancestry of the allotetraploid, A. hypogaea.
Systematic implication
The monograph of Krapovickas and Gregory (1994) for the genus Arachis is the only comprehensive systematic treatment. In their intuitive treatment, they recognized nine sections based on morphological features of the leaf, hypocotyl, fruit, flower, and branching habit. The size of these sections varied from being monotypic, for example sect. Triseminatae, to very large, for example sect. Arachis. Krapovickas and Gregory (1994) also provided information on geographic distribution and ploidy levels for these sections. Our study is the first molecular phylogenetic treatment of the entire genus using both nuclear and plastid genomic sequences. It is evident from the phylogenetic trees based on partitioned and combined datasets that, except for sect. Caulorrhizae, sect. Extranervosae, and the monotypic sect. Triseminatae, none of the other sections appears monophyletic (Figs. 1, 2). However, it is to be noted that sect. Heteranthae was represented by one species in this study.
Section Triseminatae is monotypic and it is characterized morphologically by the apomorphy of cotyledons with veins deeply sunken on the upper surface (Krapovickas and Gregory 1994). In an attempt to generate intersectional hybrids, Gregory and Gregory (1979) did not succeed in obtaining hybrids between A. triseminata and members of other sections. Further, very few hybrids were obtained when crosses were made among various accessions of A. triseminata, indicating the presence of an intraspecific barrier within this species. The two accessions of A. triseminata (GK 12881 and GK 12922) used in this study appeared in one clade with full PP and BS support (Figs. 1, 2), validating the monophyly of the section.
Similarly, this study also confirmed the monophyletic nature of sect. Caulorrhizae with both A. pintoi and A. repens appearing in one clade in all analyses with high or full BS and PP support (Figs. 1, 2). Crosses between A. repens and A. pintoi produced hybrids that had high pollen stainability (86.8%; Gregory and Gregory 1979), implying a very close genetic relationship. Similar conclusions have emerged from studies based on AFLP, microsatellite, and RAPD data (Gimenes et al. 2002b; Hoshino et al. 2006; Palmieri et al. 2005). Section Caulorrhizae is characterized by the procumbent branching habit, standard of the petals with red lines on its upper face, flowers and fruit that develop along the length of the branch, and stems that form roots at the nodes (Krapovickas and Gregory 1994). Its two species can be separated morphologically on the basis of leaf shape and leaflet size (Gimenes et al. 2002b; Krapovickas and Gregory 1994). However, despite the differences in leaf morphology, the trnT–trnF and ITS sequences were identical. Similarly, protein profiles in the Bertozo and Valls (2001) study could not discriminate between these two species.
Section Rhizomatosae (R1 and R2 genomes) as circumscribed by Krapovickas and Gregory (1994) comprises four species that are well characterized by possession of rhizomes. In this study, the section is represented by A. glabrata (R2) and A. burkartii (R1) in the ITS extended dataset (Fig. 2). The MP and BI analyses of the extended ITS dataset did not support the monophyly of the section. Arachis burkartii diverged after sect. Caulorrhizae (C genome), whereas A. glabrata was nested deeply in the erectoides clade (Fig. 2). Arachis burkartii is a diploid while A. glabrata is an autotetraploid, similar to the other two species in the section (Fernández and Krapovickas 1994; Peñaloza and Valls 2005). There is very little geographic overlap between the diploid A. burkartii and the three tetraploid species of the section (Krapovickas and Gregory 1994). Furthermore, A. burkartii differs morphologically from the remaining species by the orange standard with red veins on both surfaces and coriaceous leaves. It has been suggested, on the basis of cytogenetics, RAPD, and microsatellite markers, that the diploid and tetraploid species of sect. Rhizomatosae may not be closely related (Gregory and Gregory 1979; Stalker and Moss 1987; Angelici et al. 2008; Gimenes et al. 2002b; Nóbile et al. 2004). Although the tetraploid species placed by Krapovickas and Gregory (1994) in this section were represented in this study by only one accession of A. glabrata, it seems that the diploid A. burkartii needs further evaluation and should possibly be placed in a separate section. Consequently, our results do not support the proposed origin of the tetraploids of sect. Rhizomatosae from the diploid A. burkartii, which is in agreement with the conclusions reached by Nóbile et al. (2004).
Krapovickas and Gregory (1994) recognized sect. Extranervosae on the basis of perennial habit, expanded corolla, and orange standard with red veins on its lower surface. Our study included four of the nine species placed in this section. It is evident from our extended ITS analyses that the section is monophyletic (Fig. 2). Our results are consistent with those of Gimenes et al. (2002b), who placed A. macedoi and A. burchellii (the only two species used of sect. Extranervosae) in one cluster with high BS support, and with those of Galgaro et al. (1998) where five of the species appeared in one cluster in their UPGMA analyses of RAPD and RFLP data.
Because sect. Heteranthae (Am genome) is represented in this study by one species only, A. dardani, its taxonomic status cannot be rigorously assessed at this time. However, A. dardani seems to have phylogenetic affinity with sections Erectoides (E2), Procumbentes (E3), and Trierectoides (E1), as is evident from the analyses of partitioned and combined datasets (Figs. 1, 2). Gimenes et al. (2002b) showed A. dardani and A. rigonii of sect. Procumbentes forming a cluster in their AFLP based UPGMA phenogram, although the sampling of that study was focused on sect. Arachis with limited representation from other sections.
Most of the species representing sections Erectoides, Trierectoides, and Procumbentes in our study were formerly placed in sect. Erectoides as series Tetrafoliolatae, Trifoliolatae, and Procumbensae (Gregory et al. 1973). However, Krapovickas and Gregory (1994) raised series Procumbensae and Trifoliolatae to sectional level, whereas series Tetrafoliolatae were maintained under sect. Erectoides. Taxonomic treatment for these sections was based on plant habit, leaflet number, flower color, and hypocotyl characters (Krapovickas and Gregory 1994). Sections Erectoides and Procumbentes contain 14 and nine species, respectively, and are characterized by the tetrafoliate leaves, cylindrical hypocotyl and standard with red lines on the upper face. The two sections differ in their branching habit (erect for sect. Erectoides vs. procumbent for sect. Procumbentes) and flower position—the flowers are distributed along the branches in sect. Procumbentes and found closer to the base (collar) in sect. Erectoides. Section Trierectoides, encompassing A. guaranitica and A. tuberosa, is characterized by the trifoliate leaf and tuberiform hypocotyl. Our study demonstrates the polyphyly of all three sections and the potential of treating the whole lineage as a new taxonomic unit (Figs. 1, 2).
In a study examining intersectional relationships using crossability data, Gregory and Gregory (1979) produced hybrids among species from these three sections; however, most of these hybrids were highly or completely sterile, as indicated by pollen stainability. Krapovickas and Gregory (1994) used these cytogenetic results to support establishment of the sections Erectoides, Procumbentes, and Trierectoides. Hoshino et al. (2006) generated a dendrogram based on microsatellite data that showed species of sect. Erectoides and Procumbentes intermixed in a cluster separate from another cluster containing species of sect. Trierectoides, implying a close genetic relationship between sect. Erectoides and Procumbentes. Raina and Mukai (1999) noted in their FISH study of ribosomal DNA that species of sect. Erectoides and Procumbentes have the same genome but belong to different sub-genomes as identified by Gregory and Gregory (1979). These studies lend support to the emergence of these sections in one clade in this study.
Section Arachis is defined morphologically on the basis of the vertical peg and lack of rhizomes (Krapovickas and Gregory 1994). However, they did not further subdivide the section into the currently recognized genome groups A, B, and D. Studies using molecular markers have shown that species with the A and B genomes segregate into two distinct groups (Burow et al. 2009; Gimenes et al. 2002a; Halward et al. 1991; Hilu and Stalker 1995; Tallury et al. 2005). The sole D genome species, A. glandulifera, was shown to be more closely related to the B genome species rather than to the A genome species (Tallury et al. 2005), a relationship supported in this study (Figs. 1, 2). Tentatively, we consider groups arachis I and arachis II as two separate but closely related lineages of sect. Arachis, and, as such, sect. Arachis is also in need of taxonomic revision.
Conclusions
Phylogenetic analyses of DNA sequence information from the nuclear ITS and plastid trnT–trnF regions provided a good picture of the early evolution in Arachis and resolved new major lineages. However, the taxonomic structure of this economically important genus remains to be clarified. The results of this work support the sectional treatment of Caulorrhizae, Extranervosae, and Triseminatae as determined in the monograph by Krapovickas and Gregory (1994). In contrast, sections Erectoides, Procumbentes, and Trierectoides do not seem to be monophyletic. Further phylogenetic investigation with the use of additional genomic regions and larger taxon sampling, with morphological and cytogenetic information, will be needed to refine the taxonomy and provide a detailed phylogeny of species and genome evolution.
References
Álverez I, Wendel JF (2003) Ribosomal ITS sequence and plant phylogenetic inference. Mol Phylogenet Evol 29:417–434
Angelici C, Hoshino AA, Nobile PM, Palmieri DA, Valls JFM, Gimenes MA, Lopes CR (2008) Genetic diversity in section Rhizomatosae of the genus Arachis (Fabaceae) based on microsatellite markers. Genet Mol Biol 31:79–88
Baker FK, Lutzoni FM (2002) The utility of the incongruence length difference test. Syst Biol 51:625–637
Barkley NA, Dean RE, Pittman RN, Wang ML, Holbrook CC, Pederson GA (2007) Genetic diversity of cultivated and wild-type peanuts evaluated with M13-tailed SSR markers and sequencing. Genet Res 89:93–106
Bertozo MR, Valls JFM (2001) Seed storage protein electrophoresis in Arachis pintoi and A. repens (Leguminosae) for evaluating genetic diversity. Genet Resour Crop Evol 48:121–130
Birnboim HC, Dolye J (1979) A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucl Acids Res 7:1513–1523
Borsch T, Quandt D (2009) Mutational dynamics and phylogenetic utility of non-coding plastid DNA. Plant Syst Evol Special Issue on non-coding DNA. Evolution 282:169–199
Bravo JP, Hoshino AA, Angelici C, Lopes CR, Gimenes MA (2006) Transferability and use of microsatellite markers for the genetic analysis of the germplasm of some Arachis section species of the genus Arachis. Genet Mol Biol 29:516–524
Burow M, Simpson C, Faries M, Starr J, Paterson A (2009) Molecular biogeographic study of recently described B- and A-genome Arachis species, also providing new insights into the origins of cultivated peanut. Genome 52:107–119
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Clement M, Snell Q, Walke P, Posada D, Crandall KA (2002) TCS: estimating gene genealogies. In: Parallel distributed processing symposium (Proceedings international IPDPS. Abstracts and CD-Rom), pp 184–190
Creste S, Tsai S, Valls J, Gimenes M, Lopes C (2005) Genetic characterization of Brazilian annual Arachis species from sections Arachis and Heteranthae using RAPD markers. Genet Resour Crop Evol 52:1079–1086
Darlu P, Lecointre G (2002) When does the incongruence length difference test fail? MBE 19:432–437
Dolphin K, Belshaw R, Orme CDL, Quicke DLJ (2000) Noise and incongruence: interpreting results of incongruence length difference test. Mol Phylogenet Evol 17:401–406
Farris JS, Kallersjo M, Kluge AG, Bult C (1995) Testing significance of incongruence. Cladistics 10:315–319
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Fernández A, Krapovickas A (1994) Cromosomas y evolucion en Arachis (Leguminosae). Bonplandia 8:187–220
Galgaro L, Valls JFM, Lopes CR (1997) Study of the genetic variability and similarity among and within Arachis villosulicarpa, A. pietrarellii and A. hypogaea through isoenzyme analysis. Genet Resour Crop Evol 44:9–15
Galgaro L, Lopes CR, Gimenes M, Valls JFM, Kochert G (1998) Genetic variation between several species of section Extranervosae, Caulorrhizae, Heteranthae, and Triseminatae (genus Arachis) estimated by DNA polymorphism. Genome 41:445–454
Gimenes MA, Lopes CR, Galgaro L, Valls JFM, Kochert G (2002a) RFLP analysis of genetic variation in species of section Arachis, genus Arachis (Leguminosae). Euphytica 123:421–429
Gimenes MA, Lopes CR, Valls JFM (2002b) Genetic relationships among Arachis species based on AFLP. Genet Mol Biol 25:349–353
Gimenes MA, Hoshino AA, Barbosa AV, Palmieri DA, Lopes CR (2007) Characterization and transferability of microsatellite markers of the cultivated peanut (Arachis hypogaea). BMC Pl Biol 7:9
Gregory WC, Gregory MP (1979) Exotic germ plasm of Arachis L. interspecific hybrids. J Hered 70:185–193
Gregory WC, Gregory MP, Krapovicaks A, Smith BW, Yarbrough JA (1973) Structure and genetic resources of peanuts. In: Peanuts—culture and uses. American Peanut Research and Education Association, Inc., Stillwater, pp 47–133
Gregory WC, Krapovicaks A, Gregory MP (1980) Structure, variation, evolution and classification in Arachis. In: Summerfield RJ, Bunting AH (eds) Advances in legume sciences. Royal Botanical Gardens, Kew, pp 469–481
Halward TM, Stalker HT, LaRue EA, Kochert G (1991) Genetic variation detectable with molecular markers among unadapted germ-plasm resources of cultivated peanut and related wild species. Genome 34:1013–1020
Hammer K, Arrowsmith N, Gladis T (2003) Agrobiodiversity with emphasis on plant genomics research. Naturwissenschaften 90:241–250
Hilu KW, Stalker HT (1995) Genetic relationships between peanut and wild species of Arachis sect. Arachis (Fabaceae): evidence from RAPDs. Pl Syst Evol 198:167–178
Hoshino AA, Bravo JP, Angelici CM, Barbosa AVG, Lopes CR, Gimenes MA (2006) Heterologous microsatellite primer pairs informative for the whole genus Arachis. Genet Mol Biol 29:665–675
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Johnson LA, Soltis DE (1998) Assessing congruence: empirical examples from molecular data. In: Soltis DE, Soltis PS, Doyle JJ (eds) Molecular systematics of plants II: DNA sequencing. Kluwer, Boston, pp 297–348
Jung S, Tate PL, Horn R, Kochert G, Moore K, Abbott AG (2003) The phylogenetic relationship of possible progenitors of the cultivated peanut. J Hered 94:334–340
Kelchner SA (2000) The evolution of non-coding chloroplast DNA and its application in plant systematics. Ann Mo Bot Gard 87:482–498
Kochert G, Halward T, Branch WD, Simpson CE (1991) RFLP variability in peanut (Arachis hypogaea L) cultivars and wild species. Theor Appl Genet 81:565–570
Kovarik A, Matyasek R, Lim K, Skalicka K, Koukalova B, Knapp S, Chase M, Leitch A (2004) Concerted evolution of 18-5.8-26S rDNA repeats in Nicotiana allotetraploids. Biol J Linn Soc 82:615–625
Krapovickas A, Gregory WC (1994) Taxonomía del género Arachis (Leguminosae). Bonplandia 8:1–186
Lavia GI (1998) Karyotypes of Arachis palustris and A. praecox (Section Arachis), two species with basic chromosome number x = 9. Cytologia 63:177–181
Lavin M, Pennington RT, Klitgaard BB, Sprent JI, de Lima HC, Gasson PE (2001) The dalbergioid legumes (Fabaceae): delimitation of a pantropical monophyletic clade. Am J Bot 88:503–533
Milla SR, Isleib TG, Stalker HT (2005) Taxonomic relationships among Arachis sect. Arachis species as revealed by AFLP markers. Genome 48:1–11
Moretzsohn MDC, Hopkins MS, Mitchell SE, Kretovich S, Valls JFM, Ferreira EM (2004) Genetic diversity of peanut (Arachis hypogaea L.) and its wild relatives based on the analysis of hypervariable regions of the genome. BMC Pl Biol 4:11
Müller K (2005) SeqState: primer design and sequence statistics for phylogenetic DNA data sets. Appl Bioinf 4:65–69
Müller K (2007) PRAP2—likelihood and parsimony rachet analysis, v.09
Müller J, Müller K (2004) TreeGraph: automated drawing of complex tree figures using an extensible tree description format. Mol Ecol Notes 4:786–788
Müller K, Quandt D, Müller J, Neinhuis C (2005) PhyDE, version 0.92: phylogenetic data editor
Nixon K (1999) The parsimony ratchet, a new method for rapid parsimony analysis. Cladistics 15:407–414
Nóbile PM, Gimenes MA, Valls JFM, Lopes CR (2004) Genetic variation within and among species of genus Arachis, section Rhizomatosae. Genet Resour Crop Evol 51:299–307
Palmieri DA, Bechara MD, Curi RA, Gimenes MA, Lopes CR (2005) Novel polymorphic microsatellite markers in section Caulorrhizae (Arachis, Favaceae). Mol Ecol Notes 5:77–79
Peñaloza APS, Valls JFM (1997) Contagen do número cromossômico en assos de Arachis decora (Legumonsae). In: Vega RFA, Bovi MLA, Betti JA, Voltan RBQ (eds) Simpósio Latino Slericanno de Recursos Genéticos Vegetais. IAC/Embrapa-Cenargen, Campanias, p 21
Peñaloza APS, Valls JFM (2005) Chromosome number and satellited chromosome morphology of eleven species of Arachis (Leguminosae). Bonpladia 15:65–72
Quandt D, Müller K, Huttenen S (2003) Characterisation of the chloroplast DNA psbT-H region and the influence of dvad symmestrical elements on phylogentic reconstructions. Pl Biol 5:400–410
Quandt D, Müller K, Stech M, Frahm J-P, Frey W, Hilu KW, Borsch T (2004) Molecular evolution of the chloroplast trnL–F region in land plants. Monogr Syst Bot Mo Bot Gard 98:13–37
Raina S, Mukai Y (1999) Detection of a variable number of 18S-5.8 S-26S and 5S ribosomal DNA loci by fluorescent in situ hybridization in diploid and tetraploid Arachis species. Genome 42:52–59
Seijo JG, Lavia GI, Fernandez A, Krapovickas A, Ducasse D, Moscone EA (2004) Physical mapping of the 5S and 18S-25S rRNA genes by FISH as evidence that Arachis duranensis and A. ipaensis are the wild diploid progenitors of A. hypogaea (Leguminosae). Am J Bot 91:1294–1303
Simmons MP, Ochoterena H (2000) Gaps as characters in sequence-based phylogenetic analysis. Syst Biol 49:369–381
Singh AK, Simpson CE (1994) Biosystematics and genetic resources. In: Smartt J (ed) The groundnut crop: a scientific basis for improvement. Chapman & Hall, London, pp 96–137
Smartt J, Gregory WC, Gregory MP (1978) The genomes of Arachis hypogaea L. 1. Cytogenetic studies of putative genome donors. Euphytica 27:665–675
Soltis DE, Mavrodiev EV, Doyle JJ, Rauscher J, Soltis PS (2008) ITS and ETS sequence data and phylogeny reconstruction in allopolyploids and hybrids. Syst Bot 33:7–20
Stalker HT (1981) Hybrids in the genus Arachis between sections Erectoides and Arachis. Crop Sci 21:359–362
Stalker HT (1991) A new species in section Arachis of peanuts with a D genome. Am J Bot 78:630–637
Stalker HT, Moss JP (1987) Speciation, cytogenetics, and utilization of Arachis species. Adv Agron 41:1–40
Stöver B, Müller K (2010) TreeGraph2: combining and visualizing evidence from different phylogenetic analyses. BMC Bioinf 11:7
Swofford DL (2003) PAUP*: phylogenetic analysis using parsimony (* and other methods), version 4.0, β. Sinauer Associates, Sunderland
Taberlet P, Gilley L, Pautou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of the chloroplast DNA. Pl Mol Biol 17:1105–1109
Tallury SP, Hilu KW, Milla SR, Friend SA, Alsaghir M, Stalker HT, Quandt D (2005) Genomic affinities in Arachis section Arachis (Fabaceae): molecular and cytogenetic evidence. Theor Appl Genet 111:1229–1237
Tun YT, Yamaguchi H (2007) Phylogenetic relationship of wild and cultivated Vigna (Subgenus Ceratotropis, Fabaceae) from Myanmar based on sequence variations in non-coding regions of trnT–F. Breed Sci 57:271–280
Valls JFM, Simpson CE (1994) Taxonomy, natural distribution and attributes of Arachis. In: Kerridge PC, Hardy B (eds) Biology and agronomy of forage Arachis. CIAT, California, pp 1–18
Valls JFM, Simpson CE (2005) New species of Arachis (Leguminosae) from Brazil, Paraguay and Bolivia. Bonplandia 14:35–63
Vander Stappen J, Van Campenhout S, Gama Lopez S, Volckaert G (1998) Sequencing of the internal transcribed spacer region ITS1 as a molecular tool detecting variation in the Stylosanthes guianensis species complex. Theor Appl Genet 96:869–877
Vander Stappen J, De Laet J, Gama-López S, Van Campenhout S, Volckaert G (2002) Phylogenetic analysis of Stylosanthes (Fabaceae) based on the internal transcribed spacer region (ITS) of nuclear ribosomal DNA. Pl Syst Evol 234:27–51
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic Press, San Diego, pp 315–322
Wojciechowski MF, Lavin M, Sanderson MJ (2004) A phylogeny of legumes (Leguminosae) based on analysis of the plastid matK gene resolves many well-supported subclades within the family. Am J Bot 91:1846–1862
Acknowledgements
We thank the USDA Southern Regional PI station for some of the plant material used for this study, Deborah Wiley for growing and maintaining plant samples, and Chelsea M. Black and Daniel S. Volpe for assisting in laboratory work. This work was partially funded by the Virginia Academy of Science, the American Society of Plant Taxonomists, and the Virginia Tech Graduate Research Development Program.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Friend, S.A., Quandt, D., Tallury, S.P. et al. Species, genomes, and section relationships in the genus Arachis (Fabaceae): a molecular phylogeny. Plant Syst Evol 290, 185–199 (2010). https://doi.org/10.1007/s00606-010-0360-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00606-010-0360-8