
Abstract
In this study, outcrossing rates and genetic diversity in natural populations of Nelumbo nucifera were investigated. The estimated multilocus outcrossing rate (tm) based on 28 ISSR loci was over 90%. Analysis of genetic diversity revealed that this index was high at the species level (Hs = 0.325, I = 0.514), but low within the individual study populations (Hs = 0.148, I = 0.212). Gst-B was 0.547 and Nm was 0.414. The results of AMOVA indicated that 54.6% of the variation was due to the difference between the regions and 45.4% to the variation within the region. Although the populations were predominantly outcrossing, most of the genetic diversity was attributed to geographical effects instead of their habitats because low sexual recruitment and clonal growth deeply reduced the genetic diversity within the populations. On the basis of the high tm, Gst-B and low Nm values, we recommended that any future conservation plans should include both in situ conservation and germplasm collection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Nelumbonaceae also known as the lotus family is a small family of perennial, aquatic angiosperms, which consists of the two species Nelumbo nucifera Gaertn. and Nelumbo lutea (Willd.) Pers.. The family is characterized by simple, peltate leaves, which lack stipules and are borne on the surface of the water. Nelumbo nucifera, also called the Indian or sacred lotus, is found throughout Asia and Australia, whereas N. lutea, also known as the American lotus or water chinquapin, occurs in eastern and southern North America. Nelumbo nucifera is an important aquatic economic plant, not only as a dainty and ornamental flower but also as a source of herbal medicine with strong antipyretic, cooling, astringent, and demulcent properties (Mukherjee et al. 1997; Sinha et al. 2000; Qian 2002). The species is of religious significance in SE Asia (hence the name sacred lotus) and the seeds and leaves are also eaten in this region. Wild lotus populations are an important resource for breeding of cultivated lotus.
As an insect-pollinated, aquatic, herbaceous macrophyte, N. nucifera may reproduce by sexual (seeds) and asexual (rhizomes) means. Nelumbo nucifera has solitary and bisexual flowers. The pistil matures before the stamens (‘protogyny’). The fruit is a nut of about 1.0 g at maturity. It has a hard pericarp that surrounds the seed within (Wang and Zhang 2005). In recent years, local populations of wild lotus in Central Mainland China have been greatly reduced due to the rapid development of the aquaculture industry. Nelumbo nucifera has been included in the list of endangered species in China (Dong and Zheng 2005).
Investigation of population genetic diversity and the structure of populations of this species may therefore not only illustrate the evolutionary process and mechanisms but also provide information useful for biological conservation (Schaal et al. 1991; Friedman et al. 2004). Genetic variation in a plant species is determined by its mating system and dispersal ability of pollen and seed, which affect the genetic structure and dynamics of populations within the species (Allard 1975; Tigerstedt 1984; Muona 1990). Outcrossing rates in plant populations have traditionally been estimated from polymorphism data at isozyme loci (Adams 1983). In recent years, however, several dominant markers such as RAPD, AFLP, and ISSR have been successfully developed and can be used to assay mating systems at the DNA level (Fritsch and Rieseberg 1992; Gaiotto et al. 1997; Ge and Sun 1999). Due to their dominant behavior, these markers provide less information per locus than co-dominant markers. This is particularly relevant for applications that require genotype discrimination, as in the case of outcrossing-rate estimation. Through simulation studies, Ritland and Jain (1981) demonstrated, however, that this limitation could be readily overcome by multilocus estimation using a large number of dominant markers with intermediate gene frequencies.
Inter-simple sequence repeat (ISSR) is a technique that uses repeat-anchored primers to amplify DNA sequences between two-inverted SSR (Zietkiewicz et al. 1994). ISSR markers do not require a prior knowledge of the SSR target sequence; furthermore, they are highly reproducible due to their primer length and to the high stringency achieved by the annealing temperature. ISSR markers have been found to provide highly polymorphic fingerprints (Zietkiewicz et al. 1994; Kojima et al. 1998; Bornet et al. 2002). In an earlier study (Han et al. 2007), we successfully employed ISSR to genetic diversity and clonal diversity in N. nucifera. Our earlier study of genetic diversity of N. nucifera in China was restricted to the Liangzi Lake area in Central Province of Hubei (Han et al. 2007). In the present study, genetic diversity and outcrossing rates in the species were studied in a broader geographic area encompassing the entire natural distribution of the species in Central China.
It is important to have an account of mating processes for a species in order to effectively conserve its genetic resources and to optimize genetic improvement. We investigated the relative frequencies of outcrossing versus selfing and genetic diversity in wild populations of N. nucifera with the aim of providing insights into their population genetics in order to facilitate the conservation management of this important natural resource.
Materials and methods
Study species
Nelumbo nucifera is a perennial aquatic plant that produces insect-pollinated flowers with copious quantities of pollen. In the Yangtze River drainage area, the plant flowers from April to September. Nelumbo nucifera prefers calm adlittoral habitats such as lakes, swamps, pools and rice fields. A single individual may occupy an area of 2–4 m2 while the main rhizome may reach to 3–6 m (Wang and Zhang 2005). Dispersal may occur during the gametophyte phase by means of insect-dispersed pollen, while seed dispersal is not likely to be very efficient, given the weight of seeds for the species (approximately 1.0 g effectively excluding wind transport) and their lack of dispersal structures.
Seeds of N. nucifera were collected from a natural population (LZB covers about four square kilometers) at Liangzi Lake in Hubei Province of Central China. A total of 240 individuals (a individual refers to a seed) from a random sample of 20 natural families at intervals of 10 m, each of which offered 9–21 seeds, were used for estimation of outcrossing rates. Embryos were removed from seeds and used for genomic DNA extraction.
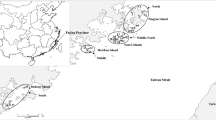
Plant materials for genetic diversity studies were obtained from ten populations with two populations each from five lake basin regions covering the entire distribution of the species in Central China (Fig. 1). The information about the ten populations is listed in Table 1. In view of the extended length of the main rhizome in N. nucifera and the expansive area occupied by a single individual of this species, sample plants for genetic diversity studies were obtained at intervals of 10 m apart, 25 sample plants were obtained from each of the ten study populations. About 5 g of young leaves from each individual plant [the term individual is used here to represent a sampling unit such as a physiologically distinct plant unit (clonal or non-clonal) at a certain distance from each other and does not necessarily denote a separate genetic individual] was obtained and placed in a zip lock plastic bag containing about 50 g of silica gel that speeded up the drying process. The samples were stored at room temperature in the laboratory.

Sampling locations of the ten study populations of N. nucifera from Central China
Genomic DNA extraction
Genomic DNA of every individual was isolated according to the protocol of Doyle and Doyle (1987). DNA was quantified by comparison with lambda DNA known concentration following electrophoresis in a 1% agarose gel using image analysis.
Evaluation of primers
Reproducibility is critical for dominant markers such as ISSR. Each primer to be tested was surveyed in duplicate reactions for each individual from the sub-sample. Only primers that yield a reproducible and clear band pattern were used. Primers used in the mating system study were screened using 16 random DNA samples of four individuals, respectively, coming from four families. Five ISSR primers were selected based on the number of polymorphisms amplified, their size, amplification intensity, reproducibility and the presence of polymorphic markers across families. In the genetic variation study, after assessing the effects of Mg2+ concentration, template DNA concentration, and temperature during the annealing stage of the amplification, 12 primers (Table 2) that produced clear and reproducible fragments were selected for further analysis.
ISSR amplification
The PCR amplification was performed in a 25 μl reaction volume containing 10 mM Tris–HCl pH 8.0, 50 mM KCl, 1.5 mM MgCl2, 0.2 mM of each dNTP, 0.4 μM of primer, 100 μg/ml of BSA, 1 unit of Taq polymerase and 30 ng of genomic DNA. Initial denaturation was for 5 min at 94°C, followed by 40 cycles of 1 min at 94°C, 1 min at specific annealing temperature, 2 min at 72°C, and a 10-min final extension step at 72°C. The annealing temperature for each primer is given in Table 2. PCR products were analyzed on 2% agarose gels at a constant voltage of 100 V for approximately 4 h, then stained with ethidium bromide, visualized with ultraviolet light and photographed.
Data analysis
The five ISSR primers amplified a total of 28 dominant markers, which were employed for the estimation of outcrossing rate. In the mating system studies, ISSR markers are typically dominant. Scoring of bands was done considering only two possible alleles: band presence or band absence. Using MLTR 3.0 (Ritland 2004), we calculated the following mating system parameters: multilocus outcrossing rate (tm), single locus outcrossing rate (ts), multilocus correlation of P (pollen and ovule gene population frequencies) estimate (rpm), single locus correlation of P estimate (rps), correlation of tm between progeny arrays (rt), and fixation index of maternal parents (F).
In the genetic variation study, ISSR amplified fragments were scored for band presence (1) or absence (0). Genetic diversity within and among populations was measured by the percentage of polymorphic bands (PPB) and Shannon’s Information index (Program POPGENE, version 1.31). Gene diversity was estimated using a Bayesian method implemented with the program HICKORY version 1.0 using all recommended default settings (Holsinger et al. 2002; Holsinger and Lewis 2003; Holsinger and Wallace 2004). HICKORY calculates the Bayesian estimate of gene diversity, Hs (average panmictic heterozygosity), by internally calculating the posterior distribution for allele frequencies at every locus in every population. The Bayesian estimator of genetic diversity is calculated for a free model and a burn-in of 50,000, a thinning sampling runs of 500,000 and factor of 100 iterations were used in each of the three replicate runs.
Hierarchical structuring of genetic variation and pairwise ΦST distances (analogous to F ST statistics at the molecular level; Excoffier et al. 1992) among the populations were determined by an analysis of molecular variance (AMOVA) with IWINAMOVA version 1.5 (Excoffier et al. 1992). Significance levels of the variance components were based on 1,000 permutations. A pairwise Euclidean distance matrix and all input files needed for the AMOVA were produced using the AMOVA PREP program version 1.01 (Miller 1998). An estimator of F ST under random-effects model of population sampling, θB, and G ST-B, a Bayesian analog of Nei’s G ST, were obtained for all populations using HICKORY (Nei 1973; Holsinger 1999; Holsinger and Lewis 2003). Cluster analysis was used to describe the genetic relationship among the different populations and the Jaccard similarity coefficients were calculated using the NTSYS-pc version 2.0 computer program package (Rohlf 1992) to generate a dendrogram via the un-weighted pair group method with arithmetic average (UPGMA). Mantel tests (Mantel 1967) were performed to analyze the effects of geographical distance on genetic variation.
Estimation bias can lead to the overestimation of parameters by as much as 5% in the dominant marker data produced by RAPD and ISSR analysis (Lynch and Milligan 1994). To reduce this bias, Lynch and Milligan proposed pruning any locus with a band frequency of higher than 1 − (3/N), where N is the number of individuals sampled. This pruning procedure was implemented with the data set prior to the genetic analyses.
Results
Mating system analysis
The five ISSR primers, including UBC 826 (7), UBC 854 (6), UBC 857 (6), UBC861 (5), and UBC 863 (4), amplified 28 dominant markers. The multilocus (tm) and single locus (ts) outcrossing estimates for the population were 0.982 ± 0.041 and 0.907 ± 0.004, respectively. The difference between the multilocus and single locus (tm − ts = 0.075 ± 0.039) was effectively zero. Multilocus correlation of P (pollen and ovule gene population frequencies) estimate (rpm), single locus correlation of P estimate (rps) and correlation of tm between progeny arrays (rt) were 0.086 ± 0.019, 0.098 ± 0.002 and 0.017 ± 0.011, respectively. Difference (rps − rpm) of estimate was 0.012 ± 0.017. The value of the single locus inbreeding coefficient of maternal parents, F, was 0.046 ± 0.021.
Genetic variation analysis
For a locus to be pruned according to Lynch and Milligan (1994) in the ISSR data (N = 250), the frequency of the band had to be 0.988 or higher. A total of 173 reproducible bands were revealed across all 250 individuals from the 10 populations. ISSR fragments from 250 to 1,600 bp were counted, with an average of 14.42 bands per applied primer.
Within populations, ISSR diversity was considerably lower than at the species level (Table 3). Bayesian gene diversity (Hs) ranged from 0.116 to 0.233, with an average of 0.148 within the populations level and 0.325 at the species level. The Shannon indices (I) ranged from 0.147 to 0.336, with an average of 0.212 within population level and 0.514 at the species level. Among the ten populations, population HLB exhibited the highest level of variability (PPB = 58.9%, Hs = 0.233, I = 0.336) and population LGB, the lowest (PPB = 25.1%, Hs = 0.116, I = 0.147). For populations in the same lake, the genetic diversity of the populations displayed a moderate increase with increase in latitude (Tables 1, 3).
Among the ten populations, a Bayesian analog of Nei’s G ST (G ST-B) was 0.547, indicating that 45.3% of the genetic diversity was within the populations. Based on G ST-B value, the mean estimated number of gene flow (Nm) between the populations was 0.414.
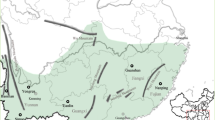
Based on Jaccard similarity coefficient, a UPGMA dendrogram among the ten populations of N. nucifera was built (Fig. 2). In the dendrogram, all the populations in each region clustered together. The populations from Diaocha Lake (DLA and DLB) and Liangzi Lake (LZA and LZB) showed the closest genetic similarity between any of the study populations. The populations from Chang Lake had less genetic similarity to those from the other four lakes. The Mantel test revealed that there was no statistically significant correlation between pairwise genetic distance and the corresponding geographic distance among the populations (r = 0.526, P = 1.000).

The unweighted pair group method with arithmetic mean (UPGMA) dendrogram for the ten populations of N. nucifera based on Jaccard similarity coefficient from ISSR markers
Analysis of molecular variance indicated that more than half of the total variations (54.6%) could be accounted for by differentiation among the five lakes, with a further 9.0% being accounted for by variation between the populations within a lake. The remainder (36.4%) was produced from individual variations within a population (Table 4). All the components of molecular variance were significant (P < 0.001).
Discussion
Dominant RAPD, AFLP, and ISSR markers were previously used to estimate the outcrossing rate in populations of flowering plants (Fritsch and Rieseberg 1992; Gaiotto et al. 1997; Ge and Sun 1999). Although ISSR technology supplies dominant markers with lower information content than traditional co-dominant isozymes, it is adequate for the study of the mating system in populations of N. nucifera. An empirical analysis by Gaiotto et al. (1997) suggested that 18 was the minimum number of dominant marker loci necessary to achieve robust estimates of tm (multilocus outcrossing rate). In our study, 28 ISSR markers loci were easily obtained with only five primers. However, this study also points to the fact that when dominant markers were applied for outcrossing rate estimation it is imperative that an adequate screening of primers should be done so as to maximize the probability of amplifying a large number of polymorphic markers in the progeny arrays.
Both multilocus (tm = 0.982 ± 0.041) and mean single locus (ts = 0.907 ± 0.004) outcrossing rates were relatively high, indicating that N. nucifera was predominantly an allogamous species. The ‘protogynous’ phenomenon (Wang and Zhang 2005) could reasonably account for the high degree of outcrossing in this species. This is to the best of our knowledge, the first published report on the outcrossing rate in N. nucifera and should contribute to optimizing genetic resources conservation and formulating breeding strategies for this species.
In the present study, the lack of any difference between the multilocus (tm) and single locus (ts) estimates indicates that there is no ‘biparental inbreeding’ in the population. In this study, the absence of ‘biparental inbreeding’ strengthens the somewhat tentative conclusion that the studied populations were predominantly outcrossing.
No differences in the outcrossing rates among the mother plants were observed [rt (correlation of tm between progeny arrays) was low]. The rp (correlation of outcrossed paternity within progeny arrays) values suggested that most of the individuals within the progeny arrays were outcrosses. The rps [single locus correlation of P (pollen and ovule gene population frequencies) estimate] was greater than rpm (multilocus correlation of P estimate), which indicated that there was an effect of population substructure on the male similarity between the outcrosses. F (fixation index of maternal parents) was near zero suggesting no Hardy–Weinberg deviations in the populations, which indicated that individuals within the populations mate at random. F in the progeny estimated was higher than that expected based on the estimate of tm. Taking tm = 0.982, the expected fixation index was [F = (1 − t)/(1 + t)] = 0.009, while the estimated F was 0.046. A higher F than the expected one suggested an excess of inbreeding and less heterozygotes than that expected in the progeny population analyzed. However, the analysis of progeny genotypes revealed that they were largely the products of outcrossing [rp (correlation of outcrossed paternity within progeny arrays) was very low]. This indicated that selfed offspring of N. nucifera did not mostly survive to reproductive maturity. The most probable mechanism causing differential survival of selfed and outcrossed progeny was partial self-incompatibility.
In the present study, the genetic diversity index [Hs (Bayesian gene diversity) = 0.325, I (the Shannon indices) = 0.514] revealed that the genetic diversity of N. nucifera was very high at the species level, but low at the within-population level (Hs = 0.148, I = 0.212). The results indicated pronounced genetic variation among the populations of the species. Similar results were found in earlier studies (Xue et al. 2006; Han et al. 2007). Geographical isolation may contribute to the high genetic diversity at the species level. The low levels of genetic diversity harbored within the study populations may be due to the following four possible reasons. Firstly, the efficient sexual recruitment was inhibited because of the unique fruit structures, which resulted in lower genetic diversity within the populations. Secondly, they might be due to low inherent variability of the ancestral species. Thirdly, the repeated decrease and increase of temperature in climatic oscillations during the Pleistocene might have caused the repeated enlarging and decreasing of populations, hence causing founder effects. This might partly account for the lower levels of variation within the populations of N. nucifera. Finally, the explosive increase in the human population and destructive utilization had caused a dramatic decline of the species. Although the populations in Longgan Lake were large in size, they had the lowest diversity owing to the destructive utilization by man.
The genetic structure of plant populations reflects the interactions of various factors, including the long-term evolutionary history of the species (shifts in distribution, habitat fragmentation and population isolation), genetic drift, mating system, gene flow and selection (Schaal et al. 1998). The founding number, probability of common origin, kin structure, and inbreeding within populations all have significant effects on genetic differentiation among populations (Whitlock and McCauley 1990). Differentiation or speciation has mainly occurred during periods when habitats were fragmented (Bridle et al. 2004). A high Gst-B (a Bayesian analog of Nei’s G ST) value (0.547) indicated pronounced genetic differentiation among the ten studied populations. AMOVA revealed that there was a considerable geographical-effected genetic variance (54.6%, P < 0.001) in N. nucifera. This was probably due to the differences in the geographic fragmentation of populations of the species. Generally, the mating system of flowering plant species greatly affects population genetic differentiation (Hamrick and Godt 1989). Estimates of genetic differentiation between the populations for outcrossing species based on AMOVA, derived by analyzing RAPD markers, have usually been <28%. For inbred species, estimates of interpopulation genetic variation have usually been >70% (Nybom and Bartish 2000). The genetic differentiation between the populations of N. nucifera (54.6%) were neither in the range of outcrossing species nor in that of inbred species. This was mainly because N. nucifera is both a predominant outcrossing and a clonal aquatic plant.
The fact that estimates of Nm (gene flow) were <1 for the species suggested that gene flow between the populations was insufficient to counter the effects of random drift (Real 1994). In this study, the relatively high genetic differentiation and low levels of gene flow detected (Nm = 0.414) strongly indicated that genetic drift had greatly affected the genetic composition of individual populations. The lack of correlation between genetic distances and geographic distances in N. nucifera suggested that there was low genetic flux between populations of this species, and that stochastic differentiation due to genetic drift had occurred. Gene flow between populations appeared to be mostly via pollen movement (Richards et al. 1999). Between-population gene flow was limited by pollen and seed dispersal. Being an insect-pollinated plant, pollen dispersal was limited by the short flight ranges of the insects. Moreover, seed dispersal is not likely to be very efficient, given the weight of seeds for the species (approximately 1.0 g effectively excluding wind transport) and their lack of dispersal structures. The limited seed dispersal contributes to the restricted gene flow and increases the probability that individuals in close physical proximity mated with one another. Both effects would promote inter-population differentiation.
Knowledge of genetic diversity within and among the populations is crucial for conservation purposes, when interpreted within a broader ecological and organismic context. Considering the high level of genetic differentiation among populations, preservation of any one population would not protect all the variation in the species. Therefore several populations throughout the entire range should be considered for conservation. If possible, all populations studied here should be conserved. But it is difficult to carry out in situ protection because of the dispersive distribution of wild populations and local continuous destroyal. The high outcrossing rate and low gene flow among the populations indicated that ex situ plantings would result in possible contamination of the different populations. Moreover, seed longevity is very long and seeds are easily preserved. Thus, germplasm collection is a better plan for conservation.
References
Adams WT (1983) Applications of isozymes in tree breeding. In: Tanksley SD, Orton TJ (eds) Isozymes in plant genetics and breeding, Part A. Elsevier Science Publishers, Amsterdam, pp 60–64
Allard RW (1975) The mating system and microevolution. Genetics 79:115–126
Bornet B, Goraguer F, Joly G, Branchard M (2002) Genetic diversity in European and Argentinean cultivated potatoes (Solanum tuberosum subsp. tuberosum) detected by inter-simple sequence repeats (ISSRs). Genome 45:481–484
Bridle JR, Pedro PM, Butlin RK (2004) Habitat fragmentation and biodiversity: testing for the evolutionary effects of refugia. Evolution 58:1394–1396
Dong YC, Zheng DS (2005) Summary of national key agriculture wild plant protection. Chinese Meteorologic Publishing Company, Beijing, p 122
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distance among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 131:479–491
Friedman WE, Moore RC, Purugganan MD (2004) The evolution of plant development. Am J Bot 91:1726–1741
Fritsch P, Rieseberg LH (1992) High outcrossing rates maintain male and hermaphrodite individuals in populations of the flowering plant Datisca glomerata. Nature 359:633–636
Gaiotto FA, Bramucci M, Grattapaglia D (1997) Estimation of outcrossing rate in a breeding population of Eucalyptus urophylla with dominant RAPD and AFLP markers. Theor Appl Genet 95:842–849
Ge XJ, Sun M (1999) Reproductive biology and genetic diversity of a crypto viviparous mangrove Aegiceras corniculatum (Myrsinaceae) using allozyme and intersimple sequence repeat (ISSR) analysis. Mol Ecol 8:2061–2069
Hamrick JL, Godt MJW (1989) Allozyme diversity in plant species. In: Brown AHD, Clegg MT, Kahler AL, Weir BS (eds) Plant population genetics, breeding and genetic resources. Sinauer, Sunderland, pp 43–63
Han YC, Teng CZ, Zhong S, Zhou MQ, Hu ZL, Song YC (2007) Genetic variation and clonal diversity in populations of Nelumbo nucifera (Nelumbonaceae) in Central China detected by ISSR markers. Aquat Bot 86:69–75
Holsinger K (1999) Analysis of genetic diversity in geographically structured populations: a Bayesian perspective. Hereditas 130:245–255
Holsinger KE, Lewis PO (2003) HICKORY: a package for analysis of population genetic data, version 1.0. Department of Ecology and Evolutionary Biology, University of Connecticut, Storrs
Holsinger KE, Wallace LE (2004) Bayesian approaches for the analysis of population genetic structure: an example from Platanthera leucophaea (Orchidaceae). Mol Ecol 13:887–894
Holsinger KE, Lewis PO, Dey DK (2002) A Bayesian approach to inferring population structure from dominant markers. Mol Ecol 11:1157–1164
Kojima T, Nagaoka T, Noda K, Ogihara Y (1998) Genetic linkage map of ISSR and RAPD markers in einkorn wheat in relation to that of RFLP markers. Theor Appl Genet 96:37–45
Lynch M, Milligan BG (1994) Analysis of population genetic structure with RAPD markers. Mol Ecol 3:91–99
Mantel NA (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Miller MP (1998) AMOVA-PREP 1.01: a program for the preparation of the AMOVA input files from dominant-marker raw data. Department of Biological Sciences, Northern Arizona University, Flagstaff, AZ
Mukherjee PK, Saha K, Das J, Pal M, Saha BP (1997) Studies on the anti-inflammatory activity of rhizomes of Nelumbo nucifera. Planta Med 63:367–369
Muona O (1990) Population genetics in forest tree improvement. In: Brown AHD, Clegg MT, Kahler AL, Weir BS (eds) Plant population genetics, breeding and genetic resources. Sinauer Press, Sunderland, MA, USA, pp 282–298
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70:3321–3323
Nybom H, Bartish IV (2000) Effects of life history traits and sampling strategies on genetic diversity estimates obtained with RAPD markers in plants. Perspect Plant Ecol Evol Syst 3(2):93–114
Qian JQ (2002) Cardiovascular pharmacological effects of bisbenzylisoquinoline alkaloid derivatives. Acta Pharmacol Sin 23:1086–1092
Real LA (1994) Ecological genetics. Princeton University Press, Princeton, NJ
Richards CM, Church S, McCauley DE (1999) The influence of population size and isolation on gene flow by pollen in Silene alba. Evolution 53:63–73
Ritland K (2004) Multilocus mating system program MLTR. http://genetics.forestry.ubc.ca/ritland/programs
Ritland K, Jain SA (1981) Model for the estimation of outcrossing rate and gene frequencies using ‘‘n’’ independent loci. Heredity 47:35–52
Rohlf FJ (1992) NTSYS–PC: numerical taxonomy and multivariate analysis system, version 2.0. State University of New York, Stony Brook, NY
Schaal BA, Hayworth DA, Olsen KM, Rauscher JT, Smith WA (1998) Phylogeographic studies in plants: problems and prospects. Mol Ecol 7:465–474
Schaal BA, Leverich WJ, Rogstad SH (1991) Comparison of methods for assessing genetic variation in plant conservation biology. In: Falk DA, Holsinger KE (eds) Genetics and conservation of rare plants. Oxford University Press, New York, pp 123–134
Sinha S, Mukherjee PK, Mukherjee K, Pal M, Mandal SC, Saha BP (2000) Evaluation of antipyretic potential of Nelumbo nucifera stalk extract. Phytother Res 14:272–274
Tigerstedt PMA (1984) Genetic mechanisms for adaptation: the mating system of Scots pine. In: Swaminathan MS, Chopra VL, Joshi BC, Sharma RP, Bansal HC (eds) Genetics: new frontiers (Proc 15th Int Congr Genetics). Oxford and IBH Publ, New Delhi, pp 317–322
Wang QC, Zhang YX (2005) Lotus varieties of China. Chinese Forestry Publishing Company, Beijing
Whitlock MC, McCauley DE (1990) Some population genetic consequences of colony formation and extinction: genetic correlations within founding groups. Evolution 44:1717–1724
Xue JH, Zhou LH, Zhou SL (2006) Genetic diversity and geographic pattern of wild lotus (Nelumbo nucifera) in Heilongjiang Province. Chin Sci Bull 51:421–432
Zietkiewicz E, Rafalski A, Labuda D (1994) Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification. Genomics 20:176–183
Acknowledgments
We thank Prof. Wang QF and Dr. Chen JM (of the School of Life Sciences, Wuhan University), Prof. Ge S and Dr. Zhu QH (of the Laboratory of Systematic and Evolutionary Botany, Institute of Botany, Chinese Academy of Sciences) for their help with data analysis. This work was supported by the National Natural Science Foundation of China (No. 30771375 and No. 30471114) and National Science and Technology Pillar Program (No. 2007BAD37B06).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Han, YC., Teng, CZ., Wahiti, G.R. et al. Mating system and genetic diversity in natural populations of Nelumbo nucifera (Nelumbonaceae) detected by ISSR markers. Plant Syst Evol 277, 13–20 (2009). https://doi.org/10.1007/s00606-008-0096-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00606-008-0096-x