
Abstract
A rapid and accurate detection of pathogens is essential for bedside or on-site diagnosis. Filter paper is an ideal diagnostic tool as it requires no equipment, possesses a high surface-area-to-volume ratio and a high capacity of capillary force. The functionalization of the surface of cellulose filter paper was explored by using glutaric anhydride, N-hydroxysuccinimide, and N,N′-dicyclohexylcarbodiimide. The activated surface systems enable aminated DNA to be immobilized on the surface of filter paper. Both synthetic oligonucleotides and bacterial genomic DNA of Staphylococcus aureus, Escherichia coli, and Campylobacter jejuni were detected successfully. The system produces a clear, consistent and highly visible brown signal within 1–5 min. The digital image can also be analyzed quantitatively due to the brown color resulting from the presence of magnetic beads. Bacterial DNA detection was accomplished by using 16S rDNA probe on the activated paper surface for universal bacterial diagnosis. The method is stable and repeatable. It can detect at least 0.5 pmol of a 120-base synthetic oligonucleotide per assay and 5–10 ng of bacterial DNA per assay.

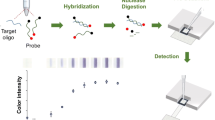
Schematic representation of the method: a. functionalization of cellulose filter paper, b. printing of aminated probes, c. incubation, d. blocking of unreacted functional groups (as dots shown), e. a visual detection of targets, f. quantitative analysis of image.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Accurate and rapid identification of pathogens is crucial for providing valuable information on the treatment and monitoring of diseases, particularly in those with rapid progression (e.g. sepsis) [1] and in cases of outbreaks (e.g. acute food-borne or water-borne diseases and nosocomial infections) [2]. Traditional culture-based diagnostic methods are time-consuming and not suitable for the fastidious pathogens. Compared to traditional culture, enzyme detection (e.g. ELISA) and nucleic acid-based diagnosis (NAD) (e.g. PCR) take less time, particularly for NAD, its high sensitivity, specificity, and capability for early diagnosis before seroconversion make it more popular [3]. However, all the methods mentioned need technical expertise and expensive equipment, which limit their applications in distant and undeveloped areas. In order to reduce mortality of infections, a rapid, cost-efficient, and user-friendly diagnostic tool is urgently required, which should also be portable and able to be operated beside patients’ beds, in physicians’ offices, at patients’ homes, or on-site of an infectious disease outbreak [4]. Efforts have been devoted to the development of biosensor systems, such as biosensor-based microfluidic systems [5,6,7]. However, electrochemical properties, such as stability and water solubility, restrict the biological usage of biosensor systems [8]. Target oligonucleotides bound to micro/nano particles (e.g. magnetic beads) are not applicable in biosensing system due to a conflict with ion current monitoring [2].
Paper-based lateral flow (immuno)assay (LFA) is a comparable technology to ELISA, which simplifies the detection method with analytes flowing through a membrane, such as pregnancy tests [9]. Since LFA is a rapid and cost-efficient method, this technology has been developed to detect a broad range of analytes including lateral flow NAD (LFNAD), relying on either immobilized antibodies or passive absorption of probe through membrane [10,11,12]. Double-stranded DNA detection (e.g. amplicons) is indirectly carried out via interactions of either antigen-antibody or avidin-biotin [13]. The basic substrate is nitrocellulose membrane in LFNAD, which is the same as in LFA [14, 15]. Low humidity makes nitrocellulose membrane difficult for handling materials due to static electricity. It is difficult for nitrocellulose membrane to handle materials due to its low humidity [9]. LFNAD has limited capability for multiplex detection and typically detects a single pathogen [16] even when a 16S probe is used [17]. Cellulose filter paper is a good alternative to nitrocellulose membrane due to its unique features: cost-efficiency, ease of use, hydrophilicity, a high capacity of capillary force, and a high surface-area-to-volume ratio [18]. Cellulose filter paper also enables multiple detection [19]. Broad-range bacterial NAD based on 16S rRNA gene is lacking in LFNAD. In LFNAD, one of the main strategies to enhance detection sensitivity is to conjugate DNA with golden particles (AuNP) since the particles possess optical properties and display variable colors based on their aggregation status [15]. When compared to golden particles, magnetic beads can be used to both purify target DNA via magnetic force and detect the DNA due to the significant contrast between white color of the filter paper and brown color of the magnetic bead [19].
In this study, based on the principle of lateral flow assay, we have developed a novel method to detect both synthetic target DNA and bacterial DNA by using functionalized cellulose filter paper and streptavidin-coated magnetic beads. Bacterial amplicons were generated with two pairs of primers targeting the 16S rRNA gene. The results show that this method can specifically identify synthetic DNA and bacterial DNA (e.g. Escherichia coli (E. coli), Staphyloccocus aureus (S. aureus), Campylobacter jejuni (C. jejuni)) in a short period of time. Since the 16S probe was used as a probe in the method, it allows a universal detection of bacteria [1, 20, 21]. The method provides a rapid, user-friendly, and instrument-free detection of bacteria.
Materials and methods
Materials and reagents
The chemicals used in this study included glutaric anhydride (GA), N-hydroxysuccinimide (NHS), N,N′-Dicyclohexylcarbodiimide (DCC), N,N-dimethylformamide (DMF), Dimethyl sulfoxide (DMSO), 3-aminopropyltriethoxysilane (APTS), SSC buffer and sodium dodecyl sulfate solution (SDS) (Sigma-Aldrich, www.sigmaaldrich.com/united-states.html). The genomic DNA of E. coli (Sigma-Aldrich), S. aureus (ATCC, www.atacc.org), C. jejuni (Sigma-Aldrich), and Saccharomyces cerevisiae (S. cerevisiae) (Sigma-Aldrich) were used. Whatman™ qualitative filter paper, DYNAL MyOne Dynabeads Streptavidin C1 (10 μg·μL−1, Fisher Scientific, www.thermofisher.com/us/en/home/brands/fisher-scientific.html), DNA oligonucleotides (Sigma-Aldrich, Table A.1), Qubit 3.0 fluorometer (Fisher Scientific) and a custom-made magnetic stand (MagRach 16, www.jenabioscience.com/proteins/purification/auxiliary-equipment/magnetic-racks-and-magnets/pp-230-magrack-16) were used as well.
Method
Selection of surface-functionalization-system of cellulose filter paper
We evaluated six different combinations of the chemicals (APTS, GA, NHS, DCC and methanol) to functionalize the surface of the filter paper.
Conversion of surface groups into carboxyl groups on filter paper
There are hydroxyl groups on cellulose surfaces [22]. Amino groups were produced by silylation with APTS (volume ratio of ethanol: H2O: APTS was 95:3:2) [23] after 24 h coating on filter paper at room temperature. The filter paper was then washed with ethanol for 5 min and deionized water for 3 min, and dried. Carboxyl groups were formed by the reaction of saturated GA in DMF with either hydroxyl or amino groups on filter paper overnight at room temperature. Then, the filter paper was washed with DMF for 5 min and deionized water for 3 min, and dried.
Formation of NHS-modified filter paper surface
Filter paper with carboxyl surface was soaked in 1 M NHS / 1 M DCC in DMF or 1 M NHS / 1 M DCC in DMSO for 4 h while shaking and then was washed with solvents (DMF or DMSO) and deionized water. In these reactions, the epoxy functional groups (-N=C=N-) in carbodimide compound (DCC) activated carboxyl groups to form an unstable intermediate, and further generated the functional groups on the surface of filter paper via the reaction between NHS and this unstable intermediate. During this process, careful washing was necessary due to the produced dicyclohexylurea byproduct. The method was labeled as GA-NHS-DCC (GND, combination 1) and APTS-GND (combination 2). Following GND and APTS-GND, filter paper was fixed with methanol (M) in both combinations overnight, and the steps of GND were repeated on both surfaces. The variants were labeled as GND-M-GND (combination 3) and APTS-GND-M-GND (combination 4). Another variant labeled M-GND (combination 5) involved fixing the surface of filter paper using methanol overnight first, followed by GND treatment. After repeating the procedure of M-GND, the surface of M-GND-M-GND (combination 6) was generated.
Printing, blocking and detection of functionalized filter paper
There were four print regions (position 1–4, Fig. S1) in each functionalized paper slide. One position was for 20 μM aminated probe DNA (APT2 or Bacterial probe), two positions were for DNA controls (20 μM unspecific aminated TID and APT2W/O without amino group modification), and the last one was for a blank control: print buffer (0.05 mM sodium phosphate, pH 8.5) (PBF). Probe DNA, control DNA and blank control were printed on the surface of filter paper manually. Then, filter paper was incubated in a humid chamber for 24 h, followed by soaking it in a blocking solution (55 °C, 50 mM ethanolamine with 100 mM Tris, pH 9.0) for 30 min, washing with 4 × SSC buffer containing 0.1% SDS for 30 min, and drying [19, 22]. During detection, activated filter paper with printed probe DNA touched a solution with target DNA bound with magnetic beads, the fluid containing target DNA transported the printed areas of the filter paper via capillary force to carry out a specific detection, followed by washing and drying. The detailed protocol was described in the section of optimization of experimental procedures based on synthetic oligonucleotides.
Image analysis
The MyImageAnalysis software was used to analyze signal intensities. The detection signals were visible on-site due to the natural brown color of magnetic beads in contrast to the white color of filter paper. The intensity of brown color was captured by software for quantitative analysis. The signal intensities were calculated by subtraction of the position 2 intensity from that of position 1. Student’s t test was applied for measuring statistical significance with a significance level set to 0.05. The background signals from position 3 and 4 were used as qualitative parameters to evaluate the optional conditions, specifically for visual detection on-site. All works were completed in triplicates under each condition.
Optimization of experimental procedures based on synthetic oligonucleotides
Evaluation of the positions of controls on activated filter paper
The aminated probe (1.5 μL of 20 μM APT2 in PBF) was always printed in position 1. The three controls (TID, APT2W/O, and PBF) were printed in position 2, 3 and 4 in different combinations. Finally, six different layouts of control positions were compared. The printed volume of each control was 1.5 μL. After incubation, the unreacted groups on the surface of filter paper were blocked. Filter paper was then washed and dried at room temperature.
Detection with synthetic target DNA bound with magnetic beads
The same volume of biotinylated target DNA (RE-APT2) and bind & wash (B&W) buffer (2 M NaCl, 1 mM EDTA, 10 mM Tris-HCl, 1 mM β-mercaptoethanol, 0.1% Tween 20, pH 7.5) were added into a tube with superparamagnetic beads coated with streptavidin. The mixture was incubated for 10 min at room temperature to form complexes of targets and beads due to the strong biological affinity of streptavidin-biotin, followed by washing with 1 × PBS-T twice. Then, the target DNA bound with beads was dissolved in 1 × PBS-T for detection [19, 22]. The initial amounts of synthetic complementary probe (APT2) and target (RE-APT2) were both 30 pmol. The initial volume of magnetic beads was 6 μL. The diameter of the magnetic beads is 1 μm.
Evaluation of wash effect
Five different wash methods after detection were compared. Three methods started with different stringent buffers — 4 × SSC buffer containing 0.01% SDS (Method 1), 2 × SSC buffer containing 0.01% SDS (Method 2), or 1 × SSC buffer containing 0.01% SDS (Method 5) — at 55 °C for 10 min, and followed by 10-fold dilution buffers — 0.4 × SSC buffer, 0.2 × SSC buffer, or 0.1 × SSC buffer — for 1 min at room temperature, respectively. Then, deionized water was used to wash the filter paper slides for 1 min at room temperature and the paper slides were dried. The other two methods involved wash only with deionized water either at 55 °C (Method 3) or at room temperature (Method 4) for 1 min.
Comparisons of solvents for printed oligonucleotides and spacer lengths
Thirty picomoles of probe and 30 pmol of each control were printed on active surface of filter paper in position 1, 2, 3, and 4. APT2 was always dissolved in water. APT2W/O and TID were dissolved in either water or Tris buffer. Six microliters of magnetic beads and 30 pmol of RE-APT2 were used in the detection on each activated filter paper. While comparing the different effect of carbon spacer (C6 and C12) at the 5′ end of printed probe, 60 pmol of probe, and 60 pmol of each control were printed on the filter paper. During the detection of each filter paper, 6 μL magnetic beads, and 60 pmol RE-APT2 were used.
Optimization of amounts of printed probe, target, and beads
Parameters, such as the amounts of printed probe, target DNA, and beads can influence the hybridization effect, and therefore need to be optimized. In order to compare the amount of printed probe, three amounts (1.5 μL, 3.0 μL, 6.0 μL of APT2) were used on the surface of functionalized filter paper, respectively. The amounts of controls were the same as that of probe on each filter paper slide. Six microliters of magnetic beads and 30 pmol of RE-APT2 were used in the detection. Sixty picomoles, 120 pmol,and 240 pmol of target DNA were compared to optimize the amount of target DNA, and the amount of printed probe and the volume of magnetic beads for each filter paper detection were 120 pmol and 6 μL. One hundred twenty picomoles of printed probe and 120 pmol of target DNA were applied to optimize the volume of magnetic beads, and 9, 12 and 15 μL of magnetic beads were compared for the detection effect on each paper slide.
Comparison of solvents for NHS/DCC
For NHS-modified filter paper surfaces, two solvents (DMSO and DMF) were used to dissolve NHS/DCC. Three concentrations of each solvent were compared (1 M, 500 mM and 250 mM). All the other steps to activate filter paper were the same as the variant of GND-M-GND. One hundred twenty picomoles of probe and controls were printed, respectively, on each active filter paper. For every paper slide detection, 6 μL of magnetic beads and 60 pmol of RE-APT2 were used.
Bacterial DNA detection
First, biotinylated PCR-products were generated and reacted with streptavidin-coated magnetic beads via the interaction between biotin and streptavidin. Both bacterial DNA and fungal DNA templates were used. The bacterial amplicons were produced via broad-range 16S rRNA gene PCR with a forward primer (64F or 530F) and a reverse primer (803R or 806R), and fungal amplicons were amplified via internal transcribed spacer 3 (ITS3) and ITS4 (Table S1). PCR reactions contained 400 nM of each primer, 1x Kapa PCR mix (Kapa Biosystems), and 0.6–0.8 ng of template in a 20 μL reaction system. Tubes were incubated in a thermocycler at 95 °C for 3 min, then cycled at 95 °C for 30 s, 50 °C for 30 s, and 72 °C for 30 s for 45 times. The concentrations of amplicons were quantitated by measuring fluorescence intensity at 520 nm with excitation at 494 nm with Qubit 3.0 fluorometer. Second, biotinylated double-stranded DNA (dsDNA) with beads was denatured into single-strand DNA (ssDNA) by 100 mM NaOH, and biotinylated ssDNA bound with beads (target ssDNA) was collected on a magnetic stand. After washing with 1 × PBS-T, target ssDNA bound with beads was dissolved in 1 × PBS-T for detection (Fig. 4) [19]. Bacterial probe was complementary to the bacterial amplicons and contained spacer sequence (Table S1).
Because of the degenerate 16S rRNA primers used in the PCRs, we compared the detection effect of filter paper, which was functionalized with different concentrations of NHS/DCC in DMSO. Briefly, 1 M of GA was used to functionalize filter paper, followed by the reactions with 250 mM, 500 mM, and 1 M of NHS/DCC in DMSO, respectively. The paper structure and functional groups were preserved with methanol, and then the reactions with GA and the same concentrations of NHS/DCC in DMSO were repeated to generate more functional groups. Around 2 μg of amplicons from the primers of 64F and 803R and 10 μL of magnetic beads were used for each slide detection. The effect of different printed volumes (6 μL, 8 μL and 10 μL) of the bacterial probe (20 μΜ) on the results of detection was also evaluated.
In order to address the limits and dynamics of detection, both synthetic bacterial target and bacterial amplicons amplified with 16S rRNA gene PCR with primers 530F and 806R were evaluated. Five concentrations of synthetic bacterial target (8.3, 83, 417, 1667 and 8333 nM) and nine concentrations of bacterial amplicons (0.08, 0.15, 0.77, 1.54, 7.7, 15.4, 30.77, 46.15, 61.54 ng·μL−1) were used, respectively. The solutions with 0 nM of bacterial target and 0 ng·μL−1 bacterial amplicons were negative controls. On each activated filter paper, 10 μL of the probe (20 μΜ) was printed, 15 μL of magnetic beads was used for the synthetic DNA detection, and 20 μL of magnetic beads was for the amplicons target.
Three different bacterial DNA genomes (E. coli, S. aureus and C. jejuni) and one fungal genome (S. cerevisiae) were used to investigate multiplicity and specificity of detection. Two micrograms of each amplicon were used on each slide with the same amounts of printed probe and beads as that in the detection limit of amplicons.
Results and discussion
In this study, we explored a new chemical surface system to functionalize filter paper for rapid and accurate DNA detection with synthetic oligonucleotides (APT2, TID, APT2W/O and PBF). The parameters were further optimized with APT2, TID, APT2W/O and PBF (e.g. print layouts of control positions, carbon spacer length, amounts of probe DNA (APT2), target DNA (RE-APT2) and magnetic beads, and wash methods after detection). Both detections of a synthetic single-stranded bacterial target and PCR-products of bacterial genomic DNA have also been explored on the activated filter paper. Every bacterial species can be detected in 1–5 min due to the universal probe and broad-range primers targeting 16S rRNA gene used. The bacterial identifications were visual on-site due to the natural color of the magnetic beads used. Furthermore, a digital image was taken with a smartphone and was quantitatively analyzed based on the captured brown color intensity in contrast to the white color filter paper by software.
Surface-activation system of cellulose filter paper
GA, DCC, and NHS can activate glass slides with functional carboxylic ester surface for nucleic acid immobilization and detection, which avoids the cross-linking phenomenon that happens to homo-bifunctional groups e.g. PDITC [23]. There is no study on the activation effect on cellulose filter paper with GA, DCC, and NHS though the filter paper possesses more space compared to glass. Considering easy accessibility and cost-effectiveness of materials, we explored the subject and discovered that the functional ester groups were also produced on the surface of filter paper (Fig. S2) and were able to be preserved via methanol. Repetition of the formation of the ester groups of NHS on the same filter paper can increase the signal intensity and homogeneity of detection (Fig. 1). The possible reason is that methanol can preserve fibers and their dimensions in filter paper and further retain the functional groups on filter paper while washing [24]. GND-M-GND was the method chosen for downstream work as it resulted in the highest signal-to-noise ratio (Fig. 1).

Comparison of the different systems to functionalize the surface of filter paper. a: five combinations (GA, NHS, DCC and methanol) were evaluated. b: comparison of two combinations based on the result of a. The scales on the Y-axis differ between graphs. Abbreviations can be found in the main text
Layout for specific probe detection on activated filter paper
The factors that interfere with the target signal intensity include physical absorption of oligonucleotides into the surface of filter paper, electrostatic repulsion between bases, and an immediate event of hybridization at room temperature (1 min - 2 min). In order to avoid cross-reactivity, we used three distinct controls, namely TID, APT2W/O and PBF. The controls of TID and APT2W/O were synthetic oligonucleotides, part of each was the same as probe of APT2, either the modification or the sequence. As the solution that the probe was dissolved in, PBF was a blank control in this study.
The strongest background was produced in position 2, so we compared the difference of intensity between position 1 and 2 under distinct situations. The best signal intensity was generated from the groups where blank control (B) was printed in position 2 instead of TID (T) and APT2W/O (A) (Fig. 2a). We assumed the unspecific oligonucleotides in position 2 would increase the background noise due to the effects of physical adsorption and oligonucleotide cross-reactivity. This hypothesis was verified by our results and the strongest noise was produced by the layout where the control of APT2WO was printed in position 2 (Fig. 2a). Significant difference in signal intensity was shown between the layouts with PBF and TID in position 2 (p = 0.009) as well as the layouts with PBF and APT2W/O in the same position (p = 0.005). There was no significant difference between the layouts with TID and APT2W/O in position 2 (p = 0.266) (Fig. 2a). We printed PBF in position 2, TID in position 3 and APT2W/O in position 4 for downstream work.

Evaluations of control position (a), wash method (b), solutions that controls dissolved in (c) and carbon spacer length (d). In figure a, A, B, T represent APT2W/O, PBF, and TID. In fig. 2b, wash methods from 1 to 5 are 4 × SSC buffer containing 0.01% SDS, 2 × SSC buffer containing 0.01% SDS, heated deionized water, room-temperature deionized water, and 1 × SSC buffer containing 0.01% SDS. The scales on the Y-axis differ among graphs
Evaluation of wash method
To provide reliable detection, it is essential to increase the ratio of signal-to-noise in diagnostic systems. It includes increasing target signal intensity, while removing the unspecific signal and decreasing the background. Optimization of wash methods is one of the options to increase signal-to-noise ratio. Comparing the different wash methods, the highest signal-to-noise ratio resulted from methods 1 and 2 (Fig. 2b, p = 0.44). The result indicated that an initial wash with a low stringent buffer is crucial to maintain hybridization signals as well as removing background [25]. Significant difference was shown between methods 1 and 5 (p = 0.014), also between 2 and 5 (p = 0.013). Therefore, we selected method 2 for subsequent work.
Evaluations of the solution printed oligonucleotides dissolved in and carbon spacer length
Tris(hydroxymethyl)aminomethane (Tris) contains a primary amine that can react with the NHS-ester groups [26] on the surface of filter paper. We compared the detection efficiency while the control ssDNA (APT2W/O and TID) was dissolved in Tris buffer and water. The result displayed that the signal intensity with controls dissolved in water was stronger than that in Tris buffer (Fig. 2c) though the difference was not significant (p = 0.3). Thus, water was chosen to dissolve the oligonucleotides in this work.
Carbon spacer can influence the hybridization efficiency since it enables maximum probe to be printed on the surface and decrease the steric hindrance of hybridization [27]. As Fig. 2d displays that a stronger hybridized signal between specific target and probe has been generated using longer carbon spacer modification at the 5′ terminal of the target DNA (p = 0.027).
Evaluations of amounts of printed probe, target and magnetic beads
In order to maximize efficacy and specificity of hybridization, we assessed different amounts of printed probe and target DNA, and different volumes of magnetic beads under the current functionalization system. The results displayed that higher amounts of printed probe, target DNA and larger volumes of bead produced stronger signal intensities (Fig. 3a, b and c). Our results verified that the porous matrix of filter paper provides larger surface area compared to the traditional platform of microarray for probe immobilization [28], and this feature enables a high amount of specific target DNA detection in a short period of time (1–2 min) at room temperature, particularly with a high amount of printed probe. The pore size of filter paper and the diameter of magnetic beads can be optimized to modify the flow rate of target solution through filter paper in order to achieve a higher ratio of signal-noise.

Optimizations of probe volume (a), target amount (b) and bead volume (c). The scales on the Y-axis differ among graphs
Evaluations of solvents dissolving NHS/DCC
It has been reported that DMSO is less toxic than DMF [29]; therefore, in the process of NHS-ester surface formation on filter paper, we compared the functionalized effect on the surface of filter paper with different solvents: DMF and DMSO, which NHS/DCC was dissolved in. During this procedure, a higher amount of dicyclohexylurea byproduct was produced in a higher concentration of NHS/DCC dissolved in a polar solvent [30]. The isourea byproduct is water insoluble [30] and makes filter paper wash difficult, specifically when the solvent was DMF. Thus, we chose 250 mM NHS/DCC in DMSO to produce NHS-ester surface of filter paper for synthetic oligonucleotides (RE-APT2) detection.
Evaluation of bacterial DNA detection
Routine diagnostic methods for detecting bacteria are based on either culture or multiple pre-designed strain-specific probe. However, many bacteria cannot be cultured and NAD assays have a limited multiplexing ability due to cross-reactivity of probe [31]. By using the detection system developed in this work, we show that activated filter paper is capable of universal detection of bacteria by targeting the 16S rRNA gene. The gene is present in all bacteria [1], and was amplified by using 16S-specific primers (Table S1). Double-stranded bacterial targets bound with beads were denatured by NaOH. Single-stranded target DNA bound with beads, which is complementary to the printed probe, was collected and purified with a magnet for the final detection (Fig. 4) [19]. This is the pilot result for the detection of a broad-range bacterial genomic DNA on an activated filter paper. Meanwhile, on-site visual signals have been achieved without wash (Movie S1). Considering that degenerate primers were used, the parameters of functionalization (e.g. concentrations of chemical solution) and detection (e.g. amount of printed probe) were optimized. The activated system with 500 mM of NHS/DCC in DMSO provided the clearest signals though there was no significant difference between the different concentrations in the effect of detection (Fig. 5a). A lower concentration of NHS/DCC (250 mM) led to a stronger background, however a higher concentration of NHS/DCC produced more byproducts (Fig. 5b) [30], which increased the difficulty in wash while the fibers were maintained. Our results also showed that a higher amount of printed probe produced a stronger intensity (Fig. 5c). Therefore, the concentration of 500 mM of NHS/DCC in DMSO and 10 μL of printed probe were chosen for synthetic single-stranded bacterial DNA and bacterial amplicon detections.

Shematic view of bacteria detection

Overview of 16S rRNA gene Detection. a. Different concentrations of NHS/DCC in DMSO were evaluated while activating filter paper. b. Visual detection of bacterial DNA on-site. c. Three volumes of printed probe were compared. The scales on the Y-axis differ between graphs
The current system can detect any bacteria due to the highly conserved 16S rRNA gene ubiquitously present in all bacteria. Three different bacterial species were detected, meanwhile bacteria signal intensity was significantly different from that of fungal genomic DNA used as control of specificity (p = 0.0007) (Fig. S3). For a 120-base synthetic bacterial target, the limit of detection (LOI) was 8.3 nM (0.5 pmol/assay). A saturated signal was shown when the concentration of the target synthetic oligonucleotide was 417 nM in the current situation (Fig. S4a). For 16S amplicons, the LOI was between 0.08 and 0.15 ng·μL−1 (between 5 and 10 ng per assay). A linear trend was displayed and the highest signal intensity was achieved with a concentration of 61.54 ng·μL−1 (R2 = 0.9533, Fig. S4b). In this work, we used degenerate primers to generate target amplicons and verified broad-range detection of bacteria on an activated filter paper system. The current method can rapidly identify bacteria, and the sensitivity of the method can potentially be further improved with advances in assay design, printing technology, and micro−/nanofluidics.
Conclusion
Cellulose filter paper is an ideal support in POC testing for DNA detection mainly due to its porous matrix. We have developed a novel surface chemistry to activate the surface of filter paper that is suitable for DNA detection in POC testing. The reactions between GA, NHS and DCC produce a carboxyl-ester surface on filter paper enabling NH2-DNA immobilization and detection. After methanol fixation on filter paper, more functional groups are generated, and thus a higher signal intensity can be achieved. As proof of concept of this activated filter paper, a rapid on-site visual detection of bacteria DNA is shown with the specific hybridization between 16S probe and 16S amplicons, which highlights the practicality and usability of the current method. The signal is visible in 1–5 min during detection, including synthetic DNA and bacterial PCR products. The method is good for rapid bacterial detection (e.g. for identification of pathogenic species instead of one single bacterium). The current method can be particularly useful in cases when multiple bacteria exist simultaneously. The method can even be integrated into a point-of-care device by combining LFA or microfluidic devices for a rapid and accurate bedside or on-site pathogen diagnostics of infectious diseases.
References
Song Y, Gyarmati P (2019) Optimized detection of bacteria in bloodstream infections. PLoS One 14:e0219086. https://doi.org/10.1371/journal.pone.0219086
Ye W, Chen T, Mao Y, Tian F, Sun P, Yang M (2017) The effect of pore size in an ultrasensitive DNA sandwich-hybridization assay for the Escherichia coli O157:H7 gene based on the use of a nanoporous alumina membrane. Microchim Acta 184(12):4435–4844
Chen Y, Wang Z, Liu Y, Wang X, Li Y, Ma P, Gu B, Li H (2018) Recent advances in rapid pathogen detection methods based on biosensors. Eur J Clin Microbial Infect Dis 37:1021–1037. https://doi.org/10.1007/s10096-018-3230-x
Dincer C, Bruch R, Kling A, Dittrich PS, Urban GA (2017) Multiplexed point-of-care testing – xPOCT. Trends Biotechnol 35:728–742. https://doi.org/10.1016/j.tibtech.2017.03.013
Arora P, Sindhu A, Dilbaghi N, Chaudhury A (2011) Biosensors as innovative tools for the detection of food borne pathogens. Biosens Bioelectron 28:1–12. https://doi.org/10.1016/j.bios.2011.06.002
Hol FJ, Dekker C (2014) Zooming in to see the bigger picture: microfluidic and nanofabrication tools to study bacteria. Science 346:1251821. https://doi.org/10.1126/science.1251821
Zhou W, Lie J, Chen Y, Cai Y, Hong Z, Chai Y (2019) Recent advances in the microfluid devices for bacteria and fungus research. TrAC Trends Anal Chem 112:175–195. https://doi.org/10.1016/j.trac.2018.12.024
Chinnadayyala SR, Park J, Le HTN, Santhosh M, Kadam AN, Cho S (2019) Recent advances in microfluidic paper-based electrochemiluminescence analytical devices for point-of-care testing applications. Biosens Bioelectron 126:68–81. https://doi.org/10.1016/j.bios.2018.10.038
Posthuma-Tumpie GA, Korf J, van Amerongen A (2009) Lateral flow (immuno)assay: its strengths, weaknesses, opportunities and threats. A literaturesurvey. Anal Bioanal Chem 393:569–582. https://doi.org/10.1007/s00216-008-2287-2
Blaskoza M, Koet M, Rauch P, Amerogen AV (2009) Development of a nucleic acid lateral flow immunoassay for simultaneous detection of Listeria spp. and Listeriamonocytogenes in food. Eur Food Res Technol 229:867–874
Carter DJ, Cary RB (2007) Lateral flow microarrays: a novel platform for rapid nucleic acid detection based on miniaturized lateral flow chromatography. Nucleic Acids Res 35:e74. https://doi.org/10.1093/nar/gkm269
Edwards KA, Baeumner EJ (2008) Liposome-enhanced lateral-flow assays for the Sandwich-hybridization detection of RNA. Methods Mol Biol 504:185–215. https://doi.org/10.1007/978-1-60327-569-9_13
Choi JR, Tang R, Wang S, Wan Abas WA, Pingguan-Murphy B, Xu F (2015) Paper-based sample-to-answer molecular diagnostic platform for point-of-care diagnostics. Biosens Bioelectron 74:427–439. https://doi.org/10.1016/j.bios.2015.06.065
Morales-Narvaez E, Naghdi T, Zor E, Merkoci A (2015) Photoluminescent lateral-flow immunoassay revealed by graphene oxide: highly sensitive paper-based pathogen detection. Anal Chem 87:8573–8577. https://doi.org/10.1021/acs.analchem.5b02383
Hu J, Wang L, Li F, Han YL, Lin M, Lu TJ, Xu F (2013) Oligonucleotide-linked gold nanoparticle aggregates for enhanced sensitivity in lateral flow assays. Lab Chip 13:4352–4357. https://doi.org/10.1039/c3lc50672j
Chen Y, Cheng N, Xu Y, Huang K, Luo Y, Xu W (2016) Point-of-care and visual detection of P. aeruginosa and its toxin genes by multiple LAMP and lateral flow nucleic acid biosensor. Biosens Bioelectron 81:317–323. https://doi.org/10.1016/j.bios.2016.03.006
Liu CC, Yeung CY, Chen PH, Yeh MK, Hou SY (2013) Salmonella detection using 16S ribosomal DNA/RNA probe-gold nanoparticles and lateral flow immunoassay. Food Chem 141:2526–2532. https://doi.org/10.1016/j.foodchem.2013.05.089
Song Y (2014) Advances in DNA detection. Dissertation, Royal Institute of Technology (KTH)
Song Y, Gyarmati P, Araujo AC, Lundeberg J, Brumer H 3rd, Stahl PL (2014) Visual detection of DNA on paper chips. Anal Chem 86:1575–1582. https://doi.org/10.1021/ac403196b
Dineva MA, MahiLum-Tapay L, Lee H (2007) Sample preparation: a challenge in the development of point-of-care nucleic acid-based assays for resource-limited settings. Analyst 132:1193–1199
Afshari A, Schrenzel J, Jeven M, Harbarth S (2012) Bench-to-bedside review: rapid molecular diagnostics for bloodstream infection – a new frontier? Crit Care 16:222. https://doi.org/10.1186/cc11202
Araújo AC, Song Y, Lundeberg J, Ståhl PL, Brumer H 3rd (2012) Activated paper surfaces for the rapid hybridization of DNA through capillary transport. Anal Chem 84:3311–3317. https://doi.org/10.1021/ac300025v
Benters R, Niemeyer CM, Drutschmann D, Blohm D, Wöhrle D (2002) DNA microarrays with PAMAM dendritic linker systems. Nucleic Acids Res 30:E10
Talbo MJ, White RG (2013) Methanol fixation of plant tissue for scanning Electron microscopy improves preservation of tissue morphology and dimensions. Plant Methods 9:36. https://doi.org/10.1186/1746-4811-9-36
Poulsen L, Soe MJ, Snakeneborg D, Moller LB, Dufva M (2008) Multi-stringency wash of partially hybridized 60-mer probes reveals that the stringency along the probe decreases with distance from the microarray surface. Nucleic Acids Res 36:E132. https://doi.org/10.1093/nar/gkn600
Savage MD (1996) An introduction to avidin-biotin technology and options for biotinylation. In: Meier T, Fahernholz F (eds) A laboratory guide to biotin-labeling in biomolecule analysis. Birkhauser Verlag, Basel, p 11
Ravan H, Kashanian S, Sanadgol N, Badoei-Dalfard A, Karami Z (2014) Strategies for optimizing DNA hybridization on surfaces. Anal Biochem 444:41–46
Xiao W, Huang J (2011) Immobilization of oligonucleotides onto zirconia-modified filter paper and specific molecularrecognition. Langmuir 27:12284–12288. https://doi.org/10.1021/la203150f
Weiss LR, Orzel RA (1967) Some comparative toxicologic and pharmacologic effects of dimethyl sulfide as a pesticide solvent. Toxicol Appl Pharmacol 11:546–557
Hermanson GT (2013) Zero-length crosslinkers. In: Hermanson GT (ed) Bioconjugate techniques, 3rd edn. Elsevier, New York, pp 259–273
Landegren U, Nilsson M (1997) Locked on target: strategies for future gene diagnostics. Ann Med 29:585–590
Acknowledgements
This study was funded by the University of Illinois College of Medicine at Peoria start-up fund to PG, and a research grant from the Rising Tide Foundation (grant number CCR-17-400) to PG.
The authors would like to express their acknowledgement to Bryan Himmel from the University of Illinois at Urbana-Champaign, for language editing.
Author information
Authors and Affiliations
Contributions
Devised and designed the study: YS and PG. Performed the experiments: YS. Analyzed the images and data: YS. Drafted the manuscript: YS. Revised and agreed upon the manuscript: YS and PG. Provided reagents: PG.
Corresponding author
Ethics declarations
The authors declare that they have no competing interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 302 kb)
(MP4 38,713 kb)
Rights and permissions
About this article

Cite this article
Song, Y., Gyarmati, P. Visual detection of bacterial DNA using activated paper stripe. Microchim Acta 186, 642 (2019). https://doi.org/10.1007/s00604-019-3748-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00604-019-3748-3