
Abstract
A high-sensitivity fluorescence immunoassay for atrazine was established. It is making use of hydrophilic NaYF4:Yb/Er upconversion nanoparticles (UCNPs) conjugated to the anti-atrazine antibody as signal probe, and of polystyrene magnetic microspheres (PMMs) conjugated to the coating antigen as the capture probe. The coating antigen on the capture probe competes with atrazine for binding to the antibody on the signal probe to form the immuno complex. The complex is separated from the test system by magnetic action, and its green fluorescence is then measured at excitation/emission wavelengths of 980/552 nm using a fluorescence spectrophotometer equipped with an external 980 nm laser. The method was applied to the determination of atrazine in corn, rice, sugar cane juice, and river water. The immunoassay has a linear range that extends from 0.005 to 10 μg·L−1. The assay also recognizes propazine and prometryn, and it therefore can be applied to detect these three herbicides simultaneously. Sugar cane juice and river water samples can be analyzed directly without any pretreatment. The detection limits for atrazine are 2 ng·L−1 in sugar cane juice and river water samples, and 20 ng·kg−1 in corn and rice samples. The recoveries of spiked samples range from 84.7 to 113.6%.

A sensitive fluorescence immunoassay combining magnetic separation for the detection of atrazine in cereal, juice, and water samples using NaYF4:Yb/Er upconversion nanoparticles as labels.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Atrazine (2-chloro-4-ethylamino-6-isopropylamino-1, 3, 5-triazine) has been used worldwide for more than 50 years. It is mainly used to effectively control the annual grasses and broad leaf weeds of corn, sugar cane and other crops by blocking the photosynthesis process of plants [1, 2]. Owing to its stable structure, long residual period and certain water solubility, atrazine will accumulate in water, soil and animals [3, 4]. Atrazine can be detected in leafy vegetables, soil and water even in urban areas. In China, the contamination of atrazine has been reported in Yangtze River Delta agricultural soils [5], and Taihu Lake [6]. Note that the trace amounts of atrazine and metabolites have great potential hazards to human health and biological system. The toxic effect of atrazine has been pointed out by a great quantity research [4]. Such as atrazine can induce endocrine disorders, affect immune regulation, cause mitochondrial dysfunction, and it also has genetic toxicity, reproductive toxicity, and carcinogenesis [7,8,9,10]. Many countries have imposed restrictions on atrazine due to its toxicological effects. Atrazine has been classified as an endocrine disrupting pesticide and its Maximum Contaminant Level (MCL) has set as 3 μg·L−1 by US Environmental Protection Agency (EPA). The International Agency for Research on Cancer (IARC) has categorized atrazine in the list of carcinogenic pesticides. The European Union (EU) has banned the use of atrazine. Considering the hazard of atrazine, it is important to establish a high sensitive method to detect atrazine.
Many methods have been developed to analyze atrazine. The conventional analytical techniques such as high-performance liquid chromatography (HPLC) [11], liquid chromatography tandem mass spectrometry (LC-MS/MS) [12], gas chromatography-mass spectrometry (GC-MS) [13], surface-enhanced Raman scattering (SERS) [14], capillary electrophoresis (CE) [15], colorimetric method [16], sequential injection chromatography (SIC) [17] have been applied to sense the trace amount atrazine in water and food samples. In addition, many immunoassays with the advantages of high sensitivity, short analysis time, specificity for target recognition, including enzyme-linked immunosorbent assay (ELISA) [18,19,20,21], immunochromatographic assay [19, 22], liposome immune lysis assay [23], chemiluminescence enzyme immunoassay [24], electrochemical immunosensor [25, 26], surface plasmon resonance immunosensor [27], fluorescent immunosensor [28] and fluorescence immunoassay [29, 30] have been proposed to detect atrazine.
Conventional downconversion fluorescent materials, such as dye, quantum dots (QDs) [28, 31], were widely used in biomedical and analytical applications due to its special optical properties. However, these materials also have many defects, such as low light transmission depth, biological toxicity, strong fluorescent background effects, potential photodamage to biological tissue, unstable chemical properties, etc. These defects will become the important factors limiting the application of down conversion materials. To address these shortcomings, alternative development of upconversion nanomaterials as fluorescent labels has attracted great attentions from researchers due to the particular luminescent properties of rare earth nanocrystals. Lanthanide-based upconversion nanoparticles (UCNPs) can emit short-wavelength light of different colors under excitation of near-infrared light. They have some advantages in comparison with traditional down conversion materials, like narrow emission peak, large Stokes shift, low toxicity, high quantum yield, and high chemical stability. Another important reason is that the luminescence of UCNPs requires near-infrared excitation, and there is no autofluorescence [32], the sensitivity can be improved to some extent. The analytical method using UCNPs as fluorescent markers to detect atrazine has not been reported so far, although there have been some reports on the detection of pesticides, antibiotics, and other targets [33, 34]. In our work, a highly sensitive fluorescence immunoassay using the coating antigen-modified polystyrene magnetic microspheres as capture probe and the antibody-modified UCNPs as signal probe was established and applied to detect atrazine in cereal, sugar cane juice, and water samples.
Experimental
Materials and apparatus
C6H9O6Yb·4H2O, C6H9O6Er·4H2O, C6H9O6Y·4H2O, diethylene glycol (DEG), polyacrylic acid (PAA), N-hydroxysuccinimide (NHS), 1-ethyl-3-[3-(dimethylamino) propyl] carbodiimide (EDC), and ovalbumin (OVA) were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA, www.sigmaaldrich.com). Oleic acid (OA, 90%), 1-octadecene (ODE, 90%), methyl alcohol, ammonium fluoride, sodium hydroxide were purchased from Sinopharm Chemical Reagent Co. Ltd. (Tianjin, China, www.sinopharmtj.cn). The anti-atrazine polyclonal antibody and atrazine-hapten were produced in our laboratory. The BCA protein quantification kits were purchased from Solarbio Science and Technology Co., Ltd. (Beijing, China, solarbio.bioon.com.cn). The magnetic microspheres were obtained from BaseLine Chrom Tech Research Centre (Tianjin, China, www.qiuhuan.com). Upconversion fluorescence intensity was measured using an F-2500 fluorescence spectrophotometer (Hitachi, Japan, www.hitachi.com) equipped with an external 0~2.4 A adjustable continuous-current 980 nm laser source (Hi-Tech Optoelectronics Co., Ltd., Beijing, China, www.htoe.cecep.cn).
Synthesis and surface modification of NaYF4:Yb, Er upconversion nanoparticles (UCNPs)
NaYF4:Yb, Er UCNPs were synthesized by reported method [34]. A total amount of 1 mmoL RE (CH3COO)3·4H2O (RE = Y, Yb, Er) with molar ratio of 78:20:2 were dissolved in a 100-mL flask containing the mixture of OA (6 mL) and ODE (17 mL). Then, the solution was stirred vigorously to make it evenly mixed at room temperature. Under the degassed state, the resulting mixture was heated to 100 °C within 10 min. The reaction mixture was ulteriorly heated to 160 °C for 30 min under the protection of an inert gas followed by cooling down to room temperature. Then a methanol solution (10 mL) containing NH4F (4 mmoL) and NaOH (2.5 mmoL) was added into the mixture and stirred for 30 min at room temperature. After that, the temperature was raised to 80 °C for 50 min in order to remove methanol. Under the degassed state, the reaction system was heated to 100 °C for 10 min. The resultant mixture was ulteriorly heated to 300 °C for 1 h. The reaction mixture was washed with ethanol and the precipitate was collected by centrifugation (10621 rcf) at 25 °C, the appeal operation was repeated three times. Finally, the OA-capped UCNPs (OA-UCNPs) were placed in a 37 °C drying oven for drying and the dried products were stored for later use.
PAA-capped UCNPs (PAA-UCNPs) were prepared by ligand exchange method [35]. Briefly, 1.5 g of PAA was added to 30 mL of diethylene glycol, and the mixture was heated to 110 °C for 1 h under vigorous agitation. Then 6 mL of toluene solution containing 90 mg of OA-UCNPs was quickly added to the above system and the resultant system was kept at 110 °C for 1 h followed by heating to 240 °C for 1 h. After cooling down to room temperature, an appropriate amount of hydrochloric acid solution was added to the above mixture for ultrasonic treatment for 10 min. The precipitate was collected by centrifugation (10621 rcf, 25 °C) for 10 min and washed three times using ultrapure water. Finally, the PAA-UCNPs were collected and placed in a 37 °C drying oven for drying and the dried products were kept in a desiccator.
Preparation of coating antigen
The coating antigen (atrazine-OVA) was prepared by an active ester method. Firstly, 4.97 mg of the atrazine-hapten (the structure of atrazine-hapten is shown in Electronic Supplementary Material, Fig. S1) was dissolved in 600 μL of anhydrous DMF by ultrasonic treatment for 5 min. Then, 17.25 mg of EDC and 10.46 mg of NHS were added into the above solution under stirring, and the mixture was continuously stirred overnight in the dark at room temperature. The resulting solution was added dropwise into 4 mL of 10 mmol·L−1 PBS (phosphate-buffered saline, 8.0 g NaCl, 0.2 g KCl, 1.42 g Na2HPO4, and 0.24 g KH2PO4 in 1 L deionized water, pH 7.4) containing 15 mg of OVA in ice-bath. Subsequently, the resulting mixture was stirred for 1 h at room temperature and then kept stirring overnight at 4 °C. Finally, the solution was dialyzed against PBS (10 mmol·L−1, pH 7.4) for three days and stored at −20 °C before use.
Preparation of probe
The active ester method was applied to prepare fluorescence signal probe. Firstly, 2.5 mg of PAA-UCNPs was dispersed in 1 mL of 2-(n-morpholino)ethanesulfonic acid buffer (MES, 10 mmol·L−1, pH 5.5) containing 2.5 mg of NHS and 5 mg of EDC by ultrasound treatment, and the mixed solution on shaking table was incubated at 30 °C for 2 h. Then the activated UCNPs were collected by centrifugation (1699 rcf) for 10 min at 4 °C and washed using ultrapure water for three times. The precipitate was added to 1 mL of N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid buffer (HEPES, 10 mmol·L−1, pH 7.2) containing serial amounts anti-atrazine antibody. The mixture was incubated at 4 °C for 12 h. The antibody-UCNPs conjugate was separated by centrifugation and washed using HEPES for three times. And then the antibody-UCNPs conjugate after final centrifugation was dissolved into 1 mL of HEPES with 10 mg of OVA and incubated at room temperature for 1 h in order to block the excess active sites on the surface of activated UCNPs. The antibody-UCNPs conjugate was centrifuged and washed using HEPES for three times. Finally, the resulting signal probe (antibody-UCNPs conjugate) was dispersed in 1 mL HEPES.
Similarly, the coating antigen can be conjugated with the carboxyl group on the surface of PMMs by active ester method to prepare the capture probe. Firstly, 5 mg of NHS and 10 mg of EDC were added to PBS (10 mmol·L−1, pH 7.4) containing 5 mg of PMMs and the mixture was incubated at room temperature for 1 h with shaking. After wash at three times with PBS, the activated PMMs were collected by magnetic separation and transferred into 1 mL of PBS. And the coating antigen was added into the above solution and incubated for 4 h. The coating antigen-PMMs conjugate was separated by external magnetic force and washed using PBS for three times. And then the coating antigen-PMMs conjugate after final magnetic separation was dissolved into 1 mL PBS containing 20 mg of OVA and incubated at room temperature for 1 h in order to block the excess active sites on the surface of activated PMMs. The coating antigen-PMMs conjugate was separated by external magnetic force and washed using PBS for three times. Finally, the resulting capture probe (coating antigen-PMMs conjugate) was dispersed in 5 mL PBS for subsequent use.
Test principle and procedure
Figure 1 displays the test principle of this fluorescence immunoassay for atrazine. In test system, the capture probe (coating antigen-PMMs conjugate) can capture the signal probe (antibody-UCNPs conjugate) to form an immune complex (PMMs-OVA-atrazine-antibody-UCNPs). The immune complex can be separated from test system by external magnetic force. In the presence of atrazine, they can compete with the capture probes to bind with the signal probes. Via 980 nm laser excitation, the fluorescence signal of the complexes at 552 nm decreases with the increase of the concentration of atrazine and there is the strongest fluorescence signal in the absence of atrazine. The use of correlation between the fluorescence signal and the concentration of atrazine allows for quantitative detection of atrazine in the sample.

Preparation process of the probes and test principle of the fluorescence immunoassay using a competitive format
First, 70 μL of signal probe and 50 μL of atrazine standard solution (or sample) were incubated in a centrifuge tube containing 50 μL of capture probe at room temperature for 50 min with shaking. The immune complex was collected via magnetic separation and washed using PBS for three times. Subsequently, the complex was dispersed in 400 μL of PBS to measure the fluorescence intensity at excitation/emission wavelengths of 980/552 nm using a fluorescence spectrophotometer (voltage: 700 V; scan speed: 1500 nm·min−1) equipped with an external 980 nm excitation source (working current: 0.9 A).
Sample treatment
The river water and fresh sugar cane juice samples can be directly measured without any treatment. For corn and rice samples, 2 mL of water/methanol extraction solution (distilled water: methanol, 1:1, v/v) was added to 1 g corn or rice sample in a centrifuge tube. The mixture was vortexed and extracted for 10 min. After centrifugation (10621 rcf, 4 °C, 10 min), the supernatant was collected and then diluted 5-fold using PBS for analysis by this method. The spiked samples with atrazine were analyzed by this method in our paper and LC-MS/MS was applied to estimate the accuracy of this method. The sample treatment process and conditions of chromatogram and mass spectrum for LC-MS/MS are provided in Electronic Supplementary Material.
Results and discussion
Characterization of UCNPs
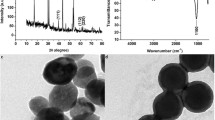
TEM images (Fig. 2a, b) show that the nanomaterials are spherical with smooth surface. Via surface modification, the formation of the hydrophilic PAA layer made the size of PAA-UCNPs relatively larger than that OA-UCNPs (see the size distribution in Fig. S2). The presence of carboxyl groups on the surface of PAA-UCNPs was confirmed by FT-IR spectroscopy. Figure 2c shows the infrared spectra of UCNPs before and after PAA treatment. Two peaks appear in 2856.4 cm−1 and 2947 cm−1, which are the symmetric and antisymmetric stretching vibration of the methylene group (-CH2) in the long chain alkyl of hydrophobic OA-UCNPs. The blunt peaks appearing at 3435 cm−1 are the characteristic peaks of the hydroxyl group (-OH). After ligand exchange using PAA, the methylene group characteristic peak disappeared and a new peak appeared at 1731.9 cm−1 in the spectrum of PAA-UCNPs. These features indicated that oleic acid on the surface of OA-UCNPs had been replaced and the carboxyl groups (-COOH) were introduced successfully on the surface of PAA-UCNPs. Figure 2d indicates the fluorescence spectra of the UCNPs with excitation at 980 nm. There was no significant difference in fluorescence features of PAA-UCNPs in comparison with OA-UCNPs, and these UCNPs had a single characteristic peak at 552 nm and no interference peaks.

Characterization of NaYF4:Yb, Er UCNPs. a, b transmission electron microscope (TEM) images, c fourier transform infrared (FT-IR) spectra, d fluorescence spectra (excitation/emission wavelengths of 980/552 nm)
Optimization of the fluorescence immunoassay
The following parameters were optimized: (a) added amount of coating antigen in preparation of capture probe; (b) added amount of antibody in preparation of signal probe; (c) added volume of signal probe; (d) incubation time. The BCA protein quantification kit can be used for detection of the amount of unconjugated coating antigen (or antibody) with the PMMs (or PAA-UCNPs). The amount of conjugated coating antigen on capture probe (or antibody on signal probe) was calculated as the difference between the total added amount of coating antigen (or antibody) and the amount of unconjugated coating antigen (or antibody). The conjugation rate expressed as a percentage (%, w/w) of the amount of conjugated coating antigen (or antibody) in the total added amount of coating antigen (or antibody).
Respective data and Figure are given in the Electronic Supplementary Material (Fig. S3). The following experimental conditions were found to give best results: (a) optimal added amount of coating antigen was 90 μg in preparation of capture probe; (b) optimal added amount of antibody was 55 μg in preparation of signal probe; (c) optimal added volume of signal probe was 70 μL in test process; (d) optimal incubation time was 50 min in test process.
Evaluation of assay sensitivity
To evaluate assay sensitivity, various concentrations of atrazine were analyzed by this fluorescence immunoassay under optimized conditions. The upconversion fluorescence intensity of test system at 552 nm with excitation at 980 nm achieves maximum in the absence of atrazine and decreases gradually as the atrazine concentration increases (Fig. 3a). There is a correlation between the concentration of atrazine and the change in fluorescence intensity (△I). Wherein △I value is calculated by the formula: ΔI = I0 − I, wherein I0 is the upconversion fluorescence intensity in the absence of atrazine, and I is the upconversion fluorescence intensity in the presence of atrazine. Figure 3b shows the change in fluorescence intensity (△I) induced by various concentrations of atrazine. The △I value increased with the increase in the concentration of atrazine and tended to changeless when the concentration of atrazine was more than 10 μg·L−1. Via plotting the △I value against the logarithmic value of each concentration, a good linearity (R2 = 0.9950) is observed in the range from 0.005 to 10 μg·L−1 with a linear equation of y = 171.75 ln(x) + 1277.2 (inset in Fig. 3b). The limit of detection (LOD) was calculated according to the 3σ rule. This fluorescence immunoassay had a 0.002 μg·L−1 of LOD for atrazine in PBS.

Sensitivity evaluation of the fluorescence immunoassay. (a) upconversion fluorescence of the immune complex at 552 nm in the absence of atrazine and in the presence of varying concentrations of atrazine with excitation at 980 nm, (b) △I values corresponding to varying concentrations of atrazine (ΔI = I0 − I, wherein I0 is the upconversion fluorescence in the absence of atrazine, and I is the upconversion fluorescence in the presence of atrazine), inset: calibration plot of the fluorescence immunoassay for atrazine in PBS. Each data point is the mean of triplicate analyses
Evaluation of assay specificity
Specificity is an important index in the evaluation of performance of detection method. The specificity of assay can be analyzed by comparing the change in fluorescence intensity (△I) in the absence and presence of atrazine and other triazine herbicides at the same concentration (10 μg·L−1). As shown in the Fig. 4, propazine and prometryn caused a same and strong fluorescence change (△I) compared with atrazine, and other structural analogues did not. Those results indicated that the fluorescence immunoassay can be used for the simultaneous detection of atrazine, propazine, and prometryn, and other triazine herbicides can not influence the performance of immunoassay.

Specificity analysis of the fluorescence immunoassay. Each data point is the mean of triplicate analyses
Sample analysis
Many compounds in food and organic reagents in sample extraction buffer may decrease the binding capability between antibody and antigen and then reduce the reliability and sensitivity of the immunoassay. This fluorescence immunoassay had good capacity of resisting interference from food matrix. The high content of sugar in juice did not influence the analytical performance of fluorescence immunoassay. No matrix affect was observed for river water and fresh sugar cane juice samples (Fig. S4), they can be directly analyzed without any pretreatment by this fluorescence immunoassay. For corn and rice samples, the sample extracted solution cannot be directly used for analysis due to the sample matrix and presence of high levels of organic reagents which can affect the specific conjugation of the antibody with antigen. Therefore, the corn and rice samples extracted solutions need to be diluted for analysis. As shown in Fig. S4, the organic reagents and sample matrix effects can be removed when the corn or rice sample extracted solution was diluted 5-fold with PBS by comparing the fluorescence intensity of test system using PBS and diluted samples extracted solution. The LODs of atrazine in river water, fresh sugar cane juice, corn and rice samples were 0.002 μg·L−1, 0.002 μg·L−1, 0.02 μg·kg−1, and 0.02 μg·kg−1, respectively.
In recoveries study, the spiked corn, rice, sugar cane juice and river water samples were analyzed simultaneously by this fluorescence immunoassay and LC-MS/MS. The recoveries of atrazine varied from 84.68% to 113.65% for this method and from 89.88% to 119.24% for LC-MS/MS (Table 1). This method has shown relatively well accuracy to detect atrazine with a correlation (R2 = 0.9931) between the results obtained by our immunoassay and LC-MS/MS. The coefficients of variation varied from 6.74% to 15.20% for this method and from 2.89% to 7.14% for LC-MS/MS. The coefficients of variation for this method were higher than those results obtained by LC-MS/MS, indicating that the precision of this fluorescence immunoassay was not as good as LC-MS/MS.
Analytical performance of the fluorescence immunoassay
Table 2 summarizes the analytical parameters of some reported immunoassays and this work. As shown in Table 2, the fluorescence immunoassay in this work displays higher sensitivity than these reported immunoassays using various detection strategies and signal label materials for the determination of atrazine. In this work, the same coating antigen (atrazine-OVA) and anti-atrazine polyclonal antibody have been applied to develop this fluorescence immunoassay and a traditional indirect competitive enzyme-linked immunosorbent assay (ELISA) simultaneously. The optimal working conditions of this traditional ELISA is summarized in Table S1 and the standard curve is shown in Fig. S5. The sensitivity (IC50) and LOD (IC15) of this traditional ELISA for atrazine in PBS are 4.28 ± 0.29 and 0.13 ± 0.04 μg·L−1, respectively. The fluorescence immunoassay is more sensitive for the detection of atrazine in PBS with a LOD of 0.002 μg·L−1. In comparison with this traditional ELISA, the fluorescence immunoassay has shown good interference-free capacity. The high content of sugar in juice do not influence the analytical performance of fluorescence immunoassay. The river water and fresh sugar cane juice samples can be directly analyzed without any pretreatment by this fluorescence immunoassay. And the river water and fresh sugar cane juice samples need to respectively be diluted 5-fold and 10-fold to eliminate matrix effect on this traditional ELISA. The LOD values of this traditional ELISA for river water and fresh sugar cane juice sample are 0.65 and 1.3 μg·L−1, respectively. Additionally, the organic reagents and sample matrix effects on the fluorescence immunoassay can be removed when the corn or rice samples extracted solution was diluted 5-fold using PBS, and 10-fold dilution can eliminate these effects on the traditional ELISA. The LOD values of corn and rice sample were 2.6 μg·kg−1. Furthermore, the fluorescence immunoassay is relatively easy to operate for test process, only one step of incubation operation and one step of washing operation are needed in whole test procedure.
Conclusion
An immunoassay is reported for sensitive detection of atrazine based on hydrophilic NaYF4:Yb/Er UCNPs with emission maximum at 552 nm as signal labels combining magnetic separation. This method is sensitive to detect atrazine in PBS with a LOD of 0.002 μg·L−1. The assay can also recognize propazine and prometryn indicating that it can be used to detect these three triazine herbicides simultaneously. This method only needs one step of incubation operation and one step of washing operation in entire test process, and the magnetic separation operation by the external magnet is more rapid and convenient. The high content of sugar in juice do not influence the analytical performance of fluorescence immunoassay and fresh sugar cane juice samples can be directly analyzed without any pretreatment. This might be due to the unique luminescence properties of upconversion nanomaterials, thereby the interference of autofluorescence is avoided effectively and the capacity of resistant to food matrix is improved. Therefore, the upconversion nanomaterials are good candidates as signal labels in analytical method system. This fluorescence immunoassay combined with magnetic separation can serve as an alternative detection approach to quantificationally detect atrazine in food and environmental samples. In this work, the added amounts of coating antigen or antibody are relatively large in preparation of capture or signal probes, thereby it causes waste of coating antigen or antibody and increase of cost. The less added amounts of coating antigen or antibody can be used for preparation of capture or signal probes in order to decrease test cost in the future.
References
Iriel A, Novo JM, Cordon GB, Lagorio MG (2014) Atrazine and methyl viologen effects on chlorophyll-a fluorescence revisited-implications in photosystems emission and ecotoxicity assessment. Photochem Photobiol 90:107–112
Czaplicka M, Barchanska H, Jaworek K, Kaczmarczyk B (2018) The interaction between atrazine and the mineral horizon of soil: a spectroscopic study. J Soils Sediments 18:827–834
Nödler K, Licha T, Voutsa D (2013) Twenty years later--atrazine concentrations in selected coastal waters of the Mediterranean and the Baltic Sea. Mar Pollut Bull 70:112–118
Singh S, Kumar V, Chauhan A, Datta S, Wani AB, Singh N, Singh J (2018) Toxicity, degradation and analysis of the herbicide atrazine. Environ Chem Lett 16:211–237
Sun JT, Pan LL, Zhan Y, Tsang DCW, Zhu LZ, Li XD (2017) Atrazine contamination in agricultural soils from the Yangtze River Delta of China and associated health risks. Environ Geochem Health 39:369–378
Ta N, Zhou F, Gao ZQ, Zhong M, Sun C (2006) The status of pesticide residues in the drinking water sources in Meiliangwan Bay, Taihu Lake of China. Environ Monit Assess 123:351–370
Fan WQ, Yanase T, Morinaga H, Condo S, Okabe T, Nomura M, Komatsu T, Morohashi KI, Hayes TB, Takayanagi R, Nawata H (2007) Atrazine-induced aromatase expression is sf-1 dependent: implications for endocrine disruption in wildlife and reproductive cancers in humans. Environ Health Perspect 115:720–727
Zhang XF, Wang MQ, Gao SY, Ren R, Zheng J, Zhang Y (2011) Atrazine-induced apoptosis of splenocytes in BALB/C mice. BMC Med 9:117
Lin J, Zhao HS, Qin L, Li XN, Zhang C, Xia J, Li JL (2018) Atrazine triggers mitochondrial dysfunction and oxidative stress in quail (Coturnix C. coturnix) cerebrum via activating xenobiotic-sensing nuclear receptors and modulating cytochrome p450 systems. J Agric Food Chem 66:6402–6413
Weber GJ, Sepúlveda MS, Peterson SM, Lewis SS, Freeman JL (2013) Transcriptome alterations following developmental atrazine exposure in zebrafish are associated with disruption of neuroendocrine and reproductive system function, cell cycle, and carcinogenesis. Toxicol Sci 132:458–466
Jia LC, Su M, Wu XQ, Sun HW (2016) Rapid selective accelerated solvent extraction and simultaneous determination of herbicide atrazine and its metabolites in fruit by ultra high performance liquid chromatography. J Sep Sci 39:4512–4519
Hu ED, Cheng HF (2013) Rapid extraction and determination of atrazine and its degradation products from microporous mineral sorbents using microwave-assisted solvent extraction followed by ultra-HPLC-MS/MS. Microchim Acta 180:703–710
Djozan D, Ebrahimi B (2008) Preparation of new solid phase micro extraction fiber on the basis of atrazine-molecular imprinted polymer: application for GC and GC/MS screening of triazine herbicides in water, rice and onion. Anal Chim Acta 616:152–159
Rubira RJG, Camacho SA, Aoki PHB, Maximino MD, Alessio P, Martin CS, Oliveira ON Jr, Fatore FM, Paulovich FV, Constantino CJL (2014) Detection of trace levels of atrazine using surface-enhanced Raman scattering and information visualization. Colloid Polym Sci 292:2811–2820
Islam K, Chand R, Han D, Kim YS (2015) Microchip capillary electrophoresis based electroanalysis of triazine herbicides. Bull Environ Contam Toxicol 94:41–45
Liu GY, Yang X, Li TF, Yu HL, Du XW, She YX, Wang J, Wang SS, Jin F, Jin MJ, Shao H, Zheng LF, Zhang YX, Zhou P (2015) Spectrophotometric and visual detection of the herbicide atrazine by exploiting hydrogen bond-induced aggregation of melamine-modified gold nanoparticles. Microchim Acta 182:1983–1989
Infante CMC, de Pra’ Urio R, Masini JC (2011) Improving the detectability of sequential injection chromatography (sic): determination of triazines by exploiting liquid core waveguide (lcw) detection. Anal Lett 44:503–513
Kaur J, Boro RC, Wangoo N, Singh KR, Suri CR (2008) Direct hapten coated immunoassay format for the detection of atrazine and 2,4-dichlorophenoxyacetic acid herbicides. Anal Chim Acta 607:92–99
Na Y, Sheng W, Yuan M, Li LL, Liu B, Zhang Y, Wang S (2012) Enzyme-linked immunosorbent assay and immunochromatographic strip for rapid detection of atrazine in water samples. Microchim Acta 177:177–184
Sai N, Sun WJ, Wu YT, Sun Z, Yu GG, Huang GW (2016) A highly sensitive immunoassay for atrazine based on covalently linking the small molecule hapten to a urea–glutaraldehyde network on a polystyrene surface. Int Immunopharmacol 40:480–486
Qie ZW, Bai JL, Xie B, Yuan L, Song N, Peng Y, Fan XJ, Zhou HY, Chen FC, Li S, Ning BA, Gao ZX (2015) Sensitive detection of atrazine in tap water using TELISA. Analyst 140:5220–5226
Shim WB, Yang ZY, Kim JY, Choi JG, Je JH, Kang SJ, Kolosova AY, Eremin SA, Chung DH (2006) Immunochromatography using colloidal gold−antibody probe for the detection of atrazine in water samples. J Agric Food Chem 54:9728–9734
Hendrickson OD, Skopinskaya SN, Yarkov SP, Zherdev AV, Dzantiev BB (2004) Development of liposome immune lysis assay for the herbicide atrazine. J Immunoass Immunochem 25:279–294
Chouhan RS, Rana KVS, Suri CR, Thampi RK, Thakur MS (2010) Trace-level detection of atrazine using immuno-chemiluminescence: dipstick and automated flow injection analyses formats. J AOAC Int 93:28–35
Liu X, Li WJ, Li L, Yang Y, Mao LG, Peng Z (2014) A label-free electrochemical immunosensor based on gold nanoparticles for direct detection of atrazine. Sensors Actuators B Chem 191:408–414
Yang M, Zhao XB, Zheng S, Liu X, Jin BW, Li H, Gan Y (2017) A new electrochemical platform for ultrasensitive detection of atrazine based on modified self-ordered Nb2O5 nanotube arrays. J Electroanal Chem 791:17–22
Farré M, Martínez E, Ramón J, Navarro A, Radjenovic J, Mauriz E, Lechuga L, Marco MP, Barceló D (2007) Part per trillion determination of atrazine in natural water samples by a surface plasmon resonance immunosensor. Anal Bioanal Chem 388:207–214
Rodriguez-Mozaz S, Reder S, de Alda ML, Gauglitz G, Barceló D (2004) Simultaneous multi-analyte determination of estrone, isoproturon and atrazine in natural waters by the river analyser (RIANA), an optical immunosensor. Biosens Bioelectron 19:633–640
Feng J, Shan GM, Maquieira A, Koivunen ME, Guo B, Hammock BD, Kennedy IM (2003) Functionalized europium oxide nanoparticles used as a fluorescent label in an immunoassay for atrazine. Anal Chem 75:5282–5286
Seidel M, Dankbar DM, Gauglitz G (2004) A miniaturized heterogeneous fluorescence immunoassay on gold-coated nano-titer plates. Anal Bioanal Chem 379:904–912
Wegner KD, Lindén S, Jin ZW, Jennings TL, el Khoulati R, van Bergen en Henegouwen PMP, Hildebrandt N (2014) Nanobodies and nanocrystals: highly sensitive quantum dot-based homogeneous FRET immunoassay for serum-based egfr detection. Small 10:734–740
Wang M, Abbineni G, Clevenger A, Mao CB, Xu SK (2011) Upconversion nanoparticles: synthesis, surface modification and biological applications. Nanomedicine 7:710–729
Hua XD, You HJ, Luo PW, Tao ZX, Chen H, Liu FQ, Wang MH (2017) Upconversion fluorescence immunoassay for imidaclothiz by magnetic nanoparticle separation. Anal Bioanal Chem 409:6885–6892
Hu GS, Sheng W, Zhang Y, Wu XN, Wang S (2015) A novel and sensitive fluorescence immunoassay for the detection of fluoroquinolones in animal-derived foods using upconversion nanoparticles as labels. Anal Bioanal Chem 407:8487–8496
Liu CH, Wang Z, Wang XK, Li ZP (2011) Surface modification of hydrophobic NaYF4:Yb,Er upconversion nanophosphors and their applications for immunoassay. Sci China Chem 54:1292–1297
Acknowledgments
This work was supported by the Open Project Program of State Key Laboratory of Food Nutrition and Safety, Tianjin University of Science and Technology (Project No. SKLFNS-KF-201819), the Tianjin Municipal Science and Technology Commission (Project No. 16PTSYJC00130), the National Key R and D Program of China (Project No. 2016YFD0401204), and the International Science and Technology Cooperation Program of China (Project No. 2014DFR30350).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The author(s) declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 537 kb)
Rights and permissions
About this article

Cite this article
Sheng, W., Shi, Y., Ma, J. et al. Highly sensitive atrazine fluorescence immunoassay by using magnetic separation and upconversion nanoparticles as labels. Microchim Acta 186, 564 (2019). https://doi.org/10.1007/s00604-019-3667-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00604-019-3667-3