
Abstract
A label-free electrochemical aptasensor was developed for selective detection of saxitoxin (STX). It is taking advantage of target-induced conformational change of an STX-specific aptamer when it binds to the toxin. A monolayer of octadecanethiol was deposited on a gold electrode, and then coated with a film of multiwalled carbon nanotubes (MWCNTs) to which the aptamer was covalently conjugated. Methylene blue (MB) was electrostatically anchored on carboxylated MWCNTs and used as the electrochemical indicator that produced a strong differential pulse voltammetric signal in the absence of target (STX). If, however, STX binds to its aptamer, this triggers a conformational change of the aptamer and results in the establishment of a barrier for heterogeneous electron transfer. The oxidation peak current of MB, acquired at −0.27 V (vs. Ag/AgCl), linearly decreases with increasing concentrations of STX in the 0.9 and 30 nM concentration range. The detection limit is 0.38 nM. Marine toxins that maybe present along with STX do not interfere even if they have a similar chemical structure. The assay was applied to the determination of STX in mussels samples and was found to be acceptably accurate. Hence, the method introduced here provides a rapid and sensitive tool for monitoring red tide pollution.

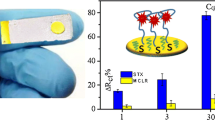
An amperometric aptasensor has been developed for STX detection in shellfish, employing capture events of aptamer-toxin and MWCNTs/SAM modified gold electrode as biosensing platform.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Saxitoxin (STX, C10H17N7O4) is one of the most harmful marine toxins which may enter the marine food chain as components of marine dinoflagellates or freshwater cyanobacterium [1], and therefore causes the so-called paralytic shellfish poisoning (PSP) via contaminated seafood or water intake [2]. Almost 2000 cases of human PSP are reported per year. PSP toxins including STX and its analogs, such as neosaxitoxin and gonyautoxins, are potent neurotoxins that can block mammalian voltage-gated sodium channels and result in death [3]. STX has been officially listed as Schedule I Chemical Warfare Agents [4]. In order to assure the food safety and the environmental sustainability, maximum permitted level (MPL) of STX at which fisheries are closed has been regulated to be 80 μg STX equivalents/100 g shellfish in most countries [5], and the monitoring programs are implemented [6, 7].
The mouse bioassay has been regulated as the reference official method for STX to provide adequately accurate assay. However, this method has received much criticism for ethical issues and other disadvantages including poor specificity, low sensitivity, and labor intensive [8]. It is gradually being replaced by chromatographic techniques, which allow separation, quantification and identification of PSP toxins [9]. Coupling techniques of liquid chromatography are approved for use by monitoring agencies, such that LC–fluorescence detection method (AOAC 2005.06) [10] is the official alternative technique in many countries, and hydrophilic interaction liquid chromatography-tandem mass spectrometry [11] can attain detection limit as low as ppb level. Apart from distinct advantages, these methods suffer from several limitations such as time-consuming sample preparation and analysis, and usage of expensive toxic standards and instruments. The European Commission encourages the use of biochemical assays and biosensors other than the official one for fast and cheap monitoring and detection of STX in multiple samples [12]. Receptor binding assay [13], in vitro cell assays [14] and immunoassays (ELISA) [4] have gradually been applied in screening of PSP toxins, and their implementation should provide an equivalent level of public health protection. The toxicity of STX and its analogs varies by approximately two orders of magnitude, due to the alternating structures of these derivatives at the four sites R1-R4 [15]. Most of the biochemical assays for STX are immunoassays based on the affinity recognition between poly- and mono-clonal antibodies and toxin, which are very sensitive and ease of use. However, potential toxin-producers often occur together in the marine system, making it very difficult to discriminate the target and its analogs accurately and determine it from sample matrices. Regarding the limitation of cross-reactivity, it is recommended that the enzyme-linked immunosorbent assay (ELISA) be used as screening tool rather than quantitative assay [16]. Biosensors have emerged as reliable alternative technique for STX quantification and monitoring. To improve the specificity of biosensors, enzymes, cell [17], artificial receptors [18] and antibody fragments have been exploited as biorecognition elements to fabricate optical, specifically based on surface plasmon resonance (SPR) [12, 19], electrochemical, or piezoelectric [20] biosensors for PSP toxins. Owing to the inherent selectivity and sensitivity of the electrochemical transduction, as well as the simple, portable and low-cost instrumentation, electrochemical biosensors have demonstrated to be the most powerful tools for in-situ detection of marine toxins.
Aptamers are single-stranded DNA or RNA oligonucleotides selected among a random oligonucleotide library that bind to a wide range of target molecules with high affinity and specificity [21]. These artificial nucleic acid ligands offer several outstanding advantages over antibodies owing to their high specificity of binding affinity, ease of synthesis, modification and storage, as well as the higher reusability, which make it more facile to transduce the capture events into detectable signals. More importantly, thanks to their three dimensional shape, aptamers can discriminate between closely related compounds on the basis of subtle structural differences. Since its pioneering application in the late 1990s, aptamers have been widely applied as biorecognition elements in electrochemical biosensors for various large molecular targets [22], mainly proteins. In the case of small target like toxin, however, the development of high performance electrochemical aptasensors remains at stagnant levels [23], due to the small changes in the interfacial-transfer resistance when it interacts with the immobilized aptamer, as well as the shortage of proper aptamers. The use of nanomaterials as labels, signal enhancers or nanostructured immobilization supports, has already been demonstrated to provide sensors with highly sensitive sensing platform. Novel transduction strategies have been incorporated in the fabrication of ochratoxin A (OTA) aptasensors to enhance electron transfer and attain higher sensitivity [12, 24]. Nanoparticles like magnetic particles or gold particles were used as electrochemical signal enhancers, offering a relatively large number of binding sites for biorecognition, and improving the sensitivity and speed of OTA biosensing.
Carbon nanotubes (CNTs) have generated great interest in electrochemical enzyme sensors and immunosensors, because of their good conductivity, electrocatalytic activity, high length-to-diameter ratio and great chemical stability [25, 26]. The unique dimensions and structure-sensitive electrical properties of the CNTs allow them to interact with some organic compounds through π-π electronic and hydrophobic interactions to form new electrical attractive nanostructures, or form a three dimensional conducting matrix that can be used for immobilization of enzyme, antibody and DNA. Much attention has been paid to the chemical functionalization and stabilization of CNTs film electrode, which will lead to the increase of accessible target sites and the enhancement of Faradaic responses [27]. Differing from other modification methods such as dip-coating and layer-by-layer assembling, controllable adsorption of CNTs onto the self assembled monolayer (SAM) of organosulfurs deposited on the gold electrode is a more precise method to obtain a stable CNTs film electrode in nanoscale [28].
In 2013, a DNA aptamer (APTSTX1) targeting STX has been selected and provided for the first time [29]. The SPR sensing results showed a selective binding of the aptamers to both surface-bound STX and free STX. Based on this ssDNA sequence, Hu et al. generated a highly specific ssDNA aptamer (F3) by SELEX, that can be used as an alternative to the STX standard and mimick STX in antibody binding [6]. Zheng et al. generated an improved method for selecting aptamer with higher affinity through rational site-directed mutation and truncation [30].
We herein present an electrochemical aptasensor for STX detection, by employing the STX aptamer (APTSTX1) that covalent immobilized onto the MWCNTs/SAM modified gold electrode as the recognition probe, together with the MB that electrostatic anchored on MWCNTs as electrochemical indicator in differential pulse voltammetry (DPV) detection. MWCNTs that controllable adsorbed on the SAM/Au electrode provides an electronically low-noise biosensing transduction platform, as well as a biocompatible interface with substantial loading ability to covalently immobilize aptamers, greatly improving the amplification and detection of capture events. In the presence of STX, target was specifically caught by DNA aptamers that immobilized on the MWCNTs/SAM modified gold electrode, thus triggered conformational changes of the aptamers and restrained electron transfer to the electrode surface, resulting in a lowered electrochemical signal of MB. The changes in the oxidation peak current of MB before and after STX recognition serve as the analytical information for sensitive and selective detection of STX. The aptasensor has been applied in the analysis of STX in real shellfish samples. It provides meaningful information for understanding the specific capture events in nanoscale substrate, which is helpful for the fabrication and application of aptasensor in toxins monitoring.
Experimental
The chemicals and apparatuses used in the experiment are detailed in the Electronic Supplementary Material (ESM).
Functionalization of MWCNTs
The carboxy - functionalized MWCNTs were prepared according to the literatures [31]. Briefly, 5 mg of MWCNTs were suspended in a mixed concentrated acid solution (H2SO4 : HNO3 = 3:1 v/v), followed by ultrasonication for 4 h at room temperature. After these treatments, the resultant suspension was centrifuged for 5 min at 7000 rpm, and then washed thoroughly with deionized water until a neutral pH was obtained. Finally, the resulting product was dried in vacuum oven overnight at 60 °C, and the collected solid was denoted as COOH-MWCNTs.
COOH-MWCNTs were further modified with methylene blue. Firstly, 0.6 mg collected COOH-MWCNTs were dispersed in 0.6 mL mixed solvent of DMF and doubly distilled water (1:1 v/v). Then, 1.2 μL of MB solution (20 mM) was well-distributed in the resultant dispersion by ultrasonication for 40 min. The MB anchored MWCNTs suspension was obtained and denoted as MB-COOH-MWCNTs.
Fabrication of aptasensor and electrochemical measurement
Prior to surface modification, the gold electrode (Au, 3 mm diameter) was polished carefully with alumina powder (1.0, 0.3 and 0.05 μm, Buehler), and then sonicated in doubly distilled water and absolute ethanol (each for 3–5 min) to remove residual alumina particles that might be trapped at the surface. The electrode was etched for 5 min in a Piranha solution (1: 3 (v/v) 30 % H2O2 and concentrated H2SO4) and then taken out of the solution and thoroughly rinsed with ultrapure Millipore water followed by ethanol. Finally, the cleanliness of the bare electrode surface was established by consecutive potential cycling in 0.5 M H2SO4 within a potential range between −0.20 and 1.60 V at a scan rate of 50 mV s−1 until a reproducible CV scan was obtained.
Gold electrode modified with SAM of C18H37SH (ODT), denoted as SAM-modified gold electrode, was prepared by immersing the pretreated gold electrode into ethanol solution of ODT (40 mM; as bifunctional linker) for 24 h at room temperature to allow the formation of a compact SAM. The electrode was then thoroughly rinsed with ethanol and doubly distilled water to remove physical adsorbed ODT, and dried with pure N2. For controllable adsorption of MB anchored MWCNTs onto the SAM-modified gold electrode, 8 μL MB-COOH-MWCNT suspension was dropped onto the surface of the SAM-modified gold electrode and followed by evaporation of the solvent in air. The resulting electrode (denoted as MB-COOH-MWCNT-SAM/Au, or MB-COOH-MWCNTs/ SAM gold electrode) was thoroughly rinsed with doubly distilled water to remove unstably adsorbed MB-COOH-MWCNTs and then dried with pure N2 before use.
Subsequently, the electrode was immersed in phosphate buffer (pH 7.0) containing 5 mM EDC and 8 mM NHS to activate the carboxy groups for 1 h, followed by further phosphate buffer rinsing. Then the electrode was immersed into the STX aptamer solution (8 μM) at room temperature for 2 h, allowing a covalent linkage of aptamers to the MWCNTs. Finally, the aptamer-modified electrode was rinsed thoroughly with Tris–HCl buffer solution to remove unbounded aptamers, and dried under a N2 stream. The resulting aptamer/MB-COOH-MWCNTs-SAM/Au electrode was then denoted as the STX aptasensor and stored at 4 °C for further use.
For cyclic voltammetry and EIS studies, each of obtained modified electrodes was transferred to the electrochemical cell containing 1 mM hexacyanoferrate and 0.1 M KCl solution.
For the detection of STX, different concentrations of standard STX solution or sample solution (5 μL) was deposited onto the electrode surface of obtained STX aptasensor and incubated for 30 min, and then carefully rinsed with Tris–HCl buffer (10 mM, pH 7.0) containing 100 mM NaCl and 2 mM MgCl2 to remove the non-binding STX. The electrochemical signals were obtained in phosphate buffer by using DPV with amplitude of 25 mV and scan rate of 20 mV s−1. The changes in the oxidation current of MB before and after the capture events were measured as a function of the STX concentration. All measurements were performed at room temperature in triplicate, and the data were obtained as mean values of three assays.
Sample preparation and extraction
The applicability of STX aptasensor was also evaluated for the analysis of STX in real samples. Different type of mussel samples, including blue mussels (sample 1) and Asia green mussels (sample 2), were randomly obtained from local supermarkets, respectively. Sample extraction was performed according to the AOAC method [32]. The outside of the mussels were thoroughly cleaned with fresh water and opened by cutting adductor muscle meat, then the inside were rinsed with distilled water to remove sand and foreign material. The mussel meats were removed carefully from shell by separating adductor muscles and tissue connecting at hinge, then draining in a sieve for 5 min to remove salt water. For representative sampling, 100 g of pooled meats were homogenized in a blender. After blending, a portion of each tissue homogenate (100 mg) of mussel meats was taken immediately and transferred accurately from the blender cup to a centrifuge tube, and used as the sub samples for further extraction. Other homogenates were stored in a tightly sealed screw-capped storage container frozen at −10 °C.
Before test, blank samples of mussel meat were collected as control samples. Sub samples were spiked with varied volume (0 , 30, 200 μL) of 0.01 mM STX standard solution respectively. 1 mL of 0.1 M HCl was added in tube and vortex mixed, boiling for 5 min. After cooling to room temperature, the acid-leached sub sample was diluted with ultrapure water to a final volume of 1 mL, and then was centrifuged at 1000 rpm for 5 min to collect the supernatant and remove the residual tissue. The sample extracts can be used for STX aptasensor assay by a 1:100 v/v dilution with Tris–HCl buffer (pH 7.0), and it should be analyzed as soon as possible. The extraction blank was obtained by performing the extraction procedure (see above) except substitute water in place of sample tissue.
Results and discussion
Principle of STX detection using electrochemical aptasensor
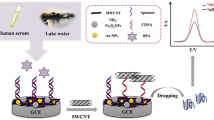
The detailed detection process of the electrochemical aptasensor can be illustrated in Scheme 1 and explained as follows. The clean gold electrode surface is first self-assembled by a dense hydrophobic monolayer of C18H37SH via the formation of Au-S bond, which isolates the electron transfer between redox solutes ([Fe(CN)6]3−/4−) and the electrode, and no electrochemical signal can be detected. Through the hydrophobic interactions with the formed SAM monolayer, MWCNTs are stably adsorbed onto the SAM-modified gold electrode in a controllable manner [33], which provides an enhanced effective loading surface for the subsequent immobilization of aptamers. More importantly, this accumulation ensures a good electrode reactivity due to the fast electron transfer between the bare Au electrode and the MWCNTs through the insulating SAM, and substantially relays the heterogeneous electron transfer between the electrode and the redox species in solution phase [28, 34]. It is reported that the secondary structure for STX aptamer (APT STX1) revealed four stem and loop segments [29], which hindered the directly assembly of aptamer-modified alkanethiol onto the gold electrode. The 3’-amino-modified STX aptamer is covalently attached to the carboxylated MWCNTs surface using the carbodiimide coupling procedure [35]. In this case, an obvious decrease of CV response current is observed, due to the fact that negative charged aptamers disturb the diffusion of the hexacyanoferrate probe to the electrode surface and partially isolate electron transfer.

The schematic diagram of amperometric aptasensor for saxitoxin using a gold electrode modified with carbon nanotubes on a self-assembled monolayer, and Methylene Blue as an electrochemical indicator probe
Owing to the well-established intercalation or binding properties of MB with guanine bases, as well as its accumulation on the MWCNTs/SAM film via the electrostatic attraction, MB is used as electrochemical indicator in the DPV sensing system (see Scheme 1). When target STX is introduced, it would bind specifically to the aptamers and form aptamer–target supramolecular complexes, during which the conformation of aptamers is dramatically changed (i.e. folding). This relatively rigid structure of folded aptamer would prevent the exposure of the bases, and accordingly restrict the electron transfer of the MB molecules that are released from the aptasensor surface. Thus, a decrease in oxidation current of MB is observed. As the change of peak current relates to the concentration of the target, an indirect detection of STX can be achieved.
Characteristics of functionalized MWCNTs
Figure 1 represents the Field Emission Scanning Electron Microscope (FE-SEM) images of COOH-MWCNTs (Fig. 1a) and the aptamer-modified COOH-MWCNTs (Fig. 1b) on the gold electrode. As can be seen, the morphology of modified MWCNTs differs in size from the well dispersed carboxyl functionalized MWCNTs. The average diameter of aptamer-immobilized COOH-MWCNTs is larger than that of COOH-MWCNTs, due to the wrapping of MWCNTs by aptamers. It may be suggested that a successful aptasensor have been prepared. An energy-dispersive X-ray spectroscopy (EDX) further confirmed the loading of aptamers on COOH-MWCNTs. Fig. S1A (ESM) showed two characteristic peaks of COOH-MWCNTs corresponding to the carbon and oxygen elements, respectively. Compared with acid-treated MWCNTs, a new characteristic peak of phosphorus at 2.035 keV can be seen obviously in Fig. S1B (ESM), which is ascribed to the presence of aptamer. The FE-SEM and EDX analysis clearly indicated that the aptamers can be covalently attached on COOH-MWCNTs successfully.

FE-SEM images of (a) COOH-MWCNT/SAM and (b) aptamer-modified COOH-MWCNT/SAM gold electrodes, and (c) the FT-IR spectra of (a) MWCNTs, (b) COOH-MWCNTs, (c) MB and (d) MB-anchored COOH-MWCNTs
To verify each step of the MWCNTs functionalization, FT-IR spectroscopy was utilized. For MWCNTs, two peaks at 3436 and 1640 cm−1 corresponded to –OH stretching vibration and the C=O stretching mode of quinone groups produced at the carbon nanotube ends, respectively (Fig. 1c, spectrum a). In the infrared spectrum of COOH-MWCNTs, the weak peak at 1720 cm−1 can be assigned to the C=O stretching vibration of carboxylic acid, and the peak at 1577 cm−1 corresponded to the stretching of C=C bond. Two peaks at 2920 and 2851 cm−1 are attributed to –CH2 asymmetrical stretching vibration and symmetrical stretching vibration (Fig. 1c, spectrum b). This indicated that carboxylic acid groups were formed on the surface of MWCNTs. After the MB conjugation, the IR spectrum of the COOH-MWCNT-MB conjugate (Fig. 1c, spectrum d) exhibited obvious characteristic absorption bands of MB, such as the ring stretching of MB at 1603 cm−1, the symmetric stretching of C-N at 1398 cm−1 and symmetric deformation of –CH3 at 1354 cm−1 (spectrum c). The strong adsorption band appeared at 1671 cm−1 indicated the C=O stretching mode of quinone groups produced at the carbon nanotube ends. These results confirmed the successful connection between COOH-MWCNTs and the MB.
Electrochemical characterizations of sensing interface
CVs of hexacyanoferrate were applied to investigate the barrier changes of the electrode surface before and after each assembly step, and to compare the electrochemical reactivity of different electrodes. A pair of well-defined redox peaks was recorded for reversible process of the redox marker at the bare Au electrode (curve a in Fig. 2a), while a tailed voltammetric response was observed for the redox system at the C18H37SH SAM/Au (curve b in Fig. 2a), indicating that electron transfer between the redox species and the Au electrode was nearly isolated by the compact SAM layer. After MWCNTs were adsorbed onto the SAM, the prepared MB-COOH-MWCNT/SAM - modified gold electrode exhibited excellent electrode reactivity without a barrier to the heterogeneous electron-transfer kinetics (curve c in Fig. 2a). The CV response is clearly dependent on the amount of COOH-MWCNTs loaded onto the SAM film. As indicated in Fig. S2 (ESM), the anodic peak current of redox marker increased with the amount of COOH-MWCNTs confined onto the SAM electrode, suggesting that the MWCNTs increase the effective electrode surface area and provide an accelerated pathway for electron transfer of the redox system. Although the interfacial capacitance of the MWCNT/SAM film electrode was slightly higher, a large amount of the MWCNTs (8 μg) was essentially needed for achieving a full surface coverage of the MWCNTs. It is obviously that the adsorption of the MWCNTs onto the SAM/Au relays the heterogeneous electron-transfer process blocked by the SAM.

a Cyclic voltammograms for (a) bare Au, (b) SAM/Au, (c) MB-COOH-MWCNT-SAM/Au, and (d) Aptamer/MB-COOH-MWCNT-SAM/Au electrodes in 0.1 M KCl containing 1.0 mM hexacyanoferrate. Scan rate: 50 mV s−1. b Nyquist diagrams for (a) bare Au, (b) MB-COOH-MWCNT-SAM/Au, (c) Aptamer/MB-COOH-MWCNT-SAM/Au, and (inset) SAM/Au electrodes at RT in 0.1 M KCl containing 1.0 mM hexacyanoferrate with the frequency range of 0.1 Hz ~ 1 MHz. Biasing potential: 0.22 V. Amplitude: 10 mV. The equivalent circuit used to fit the experimental impedance data is presented
However, the redox peak currents at the aptamer/MB-COOH-MWCNTs-SAM/Au electrode (curve d in Fig. 2a) decreased dramatically as compared to that at the MB-COOH-MWCNT-SAM/Au electrode, implying that the electron-transfer of hexacyanoferrate at solid/liquid interface was blocked. This is due to the fact that the electrostatic repulsion between anionic hexacyanoferrate and the surface-bound aptamer sequences with negative charges retards the interfacial electron-transfer kinetics. This result also demonstrates a successfully covalent immobilization of aptamers onto the MWCNTs/SAM film. EIS is a highly effective tool for monitoring the interfacial properties of electrode surface. In EIS, the diameter of semicircle at high frequencies reflects the interfacial electron transfer resistance, which controls the electron transfer kinetics of the redox marker at the electrode interface, while the linear part at low frequencies is corresponding to diffusion limited process. Figure 2b illustrated typical Nyquist plots obtained from bare Au (curve a), MB-COOH-MWCNT-SAM/Au (curve b), Aptamer/MB-COOH-MWCNT-SAM/Au (curve c) and SAM/Au (inset) electrodes at different assembly step, using [Fe(CN)6]3−/4− as the redox marker. The EIS response was analyzed by means of the Randles equivalent circuit, where Rs is the electrolyte resistance, Ret is the charge transfer resistance, Cdl is the capacitance of the electrode surface/solution interface and Zw is the Warburg impedance. As seen from curve a, the bare gold electrode exhibits a very small semicircle domain, suggesting a very low electron-transfer resistance to the redox marker in the electrolyte solution. After assembly of the ODT monolayer on the electrode surface, the SAM/Au electrode exhibited an increase of the semicircle part on the EIS spectra (inset), indicating an apparent interfacial resistance and the insulating feature of the SAM. However, further adsorption of the MWCNTs onto the SAM-modified gold electrode led to an almost straight line on the spectra of MWCNTs-SAM/Au (curve b in Fig. 2b), with a largely decreased Ret of the redox marker even at the same level of that of a bare Au, indicating that the electron transfer process was accelerated due to the presence of MWCNTs. Covalent conjugation of STX aptamer onto the MB-COOH-MWCNT-SAM modified gold electrode increases the Ret (curve c in Fig. 2b). This is attributed to the electrostatic repulsion between negatively charged phosphate groups of the aptamer and the hexacyanoferrate probe. These results were consistent with those obtained in CVs.
Improving the voltammetric response of STX aptasensor
To investigate the electrochemical response of aptasensor, CVs of the prepared aptamer/MB-COOH-MWCNT-SAM/Au electrode at different scan rates were recorded in phosphate buffer (pH 7.0) and the results are shown in Fig. 3a. A well-defined redox couple at about −0.3 V (vs. Ag/AgCl) was observed, corresponding to the binded MB. There was a good linear relationship between the peak current of MB and the scan rate from 30 to 150 mV s−1 (Fig. 3b). When the scan rate was set at 50 mV s−1, the anodic/cathodic peak current were obtained at the voltages of about −0.225/ −0.300 V (vs. Ag/AgCl), showing a peak-to-peak potential separation of about 75 mV. These results indicated that electrode process was a typical surface controlled process.

a CV responses of aptasensor in 0.1 mM phosphate buffer (pH 7.0) at scan rates (from inner to outer; a-i) of 30, 40, 50, 60, 70, 80, 90, 100 and 150 mV s−1. b The plots of redox peak current of MB obtained at about −0.3 V (vs. Ag/AgCl) vs. scan rate
Owing to the good physicochemical and electrochemical properties, MB was selected as the redox indicator for the STX aptasensor. The reproducibility of bare aptasensors prepared with different batches of STX aptamer and MB-COOH-MWCNTs was acceptable with a RSD value less than 8.5 %. As the STX added onto the aptasensor and incubated for 30 min, a specific binding between aptamers and target analyte occurred, which led to the conformational changes of aptamers, as well as the obstruction of the electron transfer of free MB in the solution to the electrode surface. Therefore, the DPV current of MB decreased with the increasing concentration of STX. The difference between the oxidation peak current intensity (ΔI) before and after STX incubation was adopted to quantitatively detect STX.
The sensing performance of aptasensor is a strong function of probe density, incubation time and buffer pH. In order to improve the detection sensitivity of the aptasensor, these parameters were carefully optimized (Fig. S3–S6, ESM). As Fig. S3 (ESM) shown, the value of ΔI increased with the increasing pH value from 6.6 to 7.0, and then decreased when pH was higher than 7.0. In order to maintain the toxicity and binding stability of STX and aptamers in applications in physiological conditions, pH value of 7.0 was chosen as the optimal pH of 0.1 mmol L−1 phosphate buffer. The amount of the surface-bound aptamer probes was studied as another factor affecting the sensing signal of the present aptasensor. From Fig. S4 (ESM), it is indicative that the sensor signal increased with the aptamer concentration till a maximum value. The poorer sensitivity of the sensor at higher concentrations of aptamer may be due to the higher steric/conformational hindrance of probe, as well as the unfavorable interactions between neighboring aptamers such as cross hybridization, reducing the response of MB as the mass transfer resistance was largely increased. The ΔI was also affected by the immobilization time of aptamer probes onto the MWCNT/SAM film, the amidation reaction between aptamers and MWCNTs was almost completed at 2 h (as shown in Fig. S5, ESM). Therefore, 7 μM aptamer covalently attached onto the MWCNT film electrode for 2 h was selected and used in throughout experiments. The incubation time for the interaction of 4.0 nM STX and the surface-bound aptamer probes was optimized (Fig. S6, ESM). The results indicate that the current response of MB first increases with incubation time, and then reaches a plateau (after 30 min) that indicates saturation.
Analytical performance of the aptasensor
To ensure applicability of the aptasensor for sensitive quantification of STX, the DPV current responses obtained at different concentrations of STX are evaluated under the optimal experimental conditions. As Fig. 4a shown, the oxidation peak current of MB observed in this sensing system gradually decreased with an increasing introduction of STX target, as a consequence of the efficient capture of STX by the aptamers.

a DPVs obtained for aptasensor after incubation with different concentrations of STX (0, 0.9, 3.0, 5.0, 7.0, 9.0, 20, 30 nM, respectively) in 0.1 mM phosphate buffer (pH 7.0). Scan rate: 20 mV s−1. Inset: the plot of relative current response of MB (ΔI) vs. STX concentration. All measurements were carried out in triplicate. b Comparison of relative current responses (ΔI) of MB in the presence of different toxins at the same concentration of 18 nM (error bars: SD, n = 3)
By analyzing the change between anodic current responses of MB and the concentrations of STX, a calibration curve by plotting the ΔI of MB before and after STX incubation and the STX concentration was presented in the inset of Fig. 4a. The results showed that the ΔI value increased linearly with STX concentration in the range of 0.9 to 30 nM (equal to 0.27–9.0 g L−1), and the following equation was found: ΔI (μA) = 0.4402 × C[STX] (nM) + 0.2907, R2 = 0.9931. The detection limit (S/N = 3) was calculated as 0.38 nM (0.11 g L−1), which is comparable or even lower than other reported detection methods for STX so far [36, 37]. Table S1 (ESM) lists the analytical performance of the aptasensor compared with some other STX sensors reported in the literatures. The relatively high sensitivity of the aptasensor is attributed to the enhanced loading of aptamers on the MWCNTs/SAM modified gold electrode, and the high binding affinity of target analyte to aptamers.
The reproducibility and stability of the aptasensor were estimated. Ten repetitive measurements of 5 nM STX solution with the aptasensor was performed under the optimized sensing conditions. Acceptable and reproducible DPV signals with relative standard deviation (RSD) of 8.1 % were obtained. The storage stability of the aptasensor was examined by determining current response toward 5 nM of STX. When the aptasensor was stored at 4 °C in dry condition, it retained about 90 % of its initial response signal after a 2 weeks storage period, indicating the good stability.
The selectivity of aptasensor for STX detection
The accurately discrimination of STX and its analogs from complex matrices is especially difficult due to the fact of co-occurrence. In order to evaluate the selectivity of the aptasensor toward STX, control experiments were performed by measuring DPV response signal of the sensing system, employing okadaic acid (OA), neoSTX and GTX1&4 as the interfering species. Figure 4b shows different current responses of aptasensor after adding 18 nM STX and three interfering toxins under the same experimental conditions. As shown in figure, compared with the small decreases of MB oxidation peak current obtained after three interfering toxins added respectively, a significant decrease in current response induced by the interaction of aptamer probe and STX was observed. This result can be ascribed to the fact that the capture of STX by aptamers is based on the specific binding but not on the other factors, such as nonspecific adsorption. Thus, the aptasensor is specific for STX over other toxins, and it therefore can be used to detect and quantify STX with high selectivity. Apart from eliminating the cross-reactivity of antibody against other toxin analogs, the use of aptasensor also avoids the time-consuming process in both preparation and package of antibody for immunosensor. Owing to the ease of use and stability, this aptasensor provides a simple and realistic sensing platform for rapid monitoring of shellfish and marine pollution.
STX detection in real samples
The aptasensing platform was then applied for the STX detection in mussel samples. As the mussels were obtained from local market, collected mussel samples are usually not polluted and do not contain toxins in most cases. To demonstrate the feasibility of the method, a recovery test was performed by spiking two different concentrations of STX into sub samples of mussels, following with the pretreatment procedure as mentioned in the section of sample preparation and extraction. Corresponding DPV current responses were then measured with the aptasensor and analyzed. As listed in Table 1, recoveries were from 63 to 121 %, indicating an acceptable accuracy and reproducibility. Moreover, this electrochemical aptasensing strategy is fast response (35 s), cost-effective and less technical demanding than many analytical methods reported so far.
Conclusions
In summary, we have developed a selective and sensitive electrochemical aptasensor for rapid determination of STX, employing a stable MWCNT/SAM film electrode as a low interfacial capacitive transduction platform and MB as a redox indicator. MWCNTs function properly as a signal amplifier to improve both the loading ability of aptamers on its surface and the electron-transfer between the redox indicator and the gold electrode. The detection of STX is achieved by measuring the change of DPV current responses of MB, which is caused by the conformational changes triggered by the specific capture events of aptamers and STX. This aptasensor using MWCNT/SAM film electrode has some advantages over the other reported sensing method with respect to sensitivity, specificity, analysis speed and cost. It is demonstrated to be a viable sensing strategy for monitoring the occurrence of PSP in marine water and seafood, and also has potential application in detection of capture events to other toxins. However, the detection linear range with the reported aptasensor is only limited within three orders of magnitude (0.9–40 nM), similar to most of other aptasensors. Future work will be focused on the application to the red tide pollution analysis with the aptasensor.
References
Deeds J, Landsberg J, Etheridge S, Pitcher G, Longan S (2008) Non-traditional vectors for paralytic shellfish poisoning. Mar Drugs 6:308–348
Etheridge S (2010) Paralytic shellfish poisoning: seafood safety and human health perspectives. Toxicon 56:108–122
Hoehne A, Behera D, Parsons W, James M, Shen B, Borgohain P, Bodapati D, Prabhakar A, Gambhir S, Yeomans D, Biswal S, Chin F, Bois J (2013) A 18F-labeled saxitoxin derivative for in vivo PET-MR imaging of voltage-gated sodium channel expression following nerve injury. J Am Chem Soc 135:18012–18015
Cusick K, Sayler G (2013) An overview on the marine neurotoxin, saxitoxin: genetics, molecular targets, methods of detection and ecological functions. Mar Drugs 11:991–1018
Van Egmond H (2004) Marine biotoxins: food and nutrition paper 80. Food and Agricultural Organization of the United Nations (FAO), Rome
Hu P, Liu Z, Tian R, Ren H, Wang X, Lin C, Gong S, Meng X, Wang G, Zhou Y, Lu S (2013) Selection and identification of a DNA aptamer that mimics saxitoxin in antibody binding. J Agric Food Chem 61:3533–3541
Fraga M, Vilariño N, Louzao M, Rodríguez P, Campbell K, Elliott C, Botana L (2013) Multidetection of paralytic, diarrheic, and amnesic shellfish toxins by an inhibition immunoassay using a microsphere-flow cytometry system. Anal Chem 85:7794–7802
Campbell K, Stewart L, Doucette G, Fodey T, Haughey S, Vilarino N, Kawatsu K, Elliott C (2007) Assessment of specific binding proteins suitable for the detection of paralytic shellfish poisons using optical biosensor technology. Anal Chem 79:5906–5914
de la Iglesia P, Barber E, Giménez G, Rodríguez-Velasco M, Villar-Donzlez A, Diogne J (2011) High-throughput analysis of amnesic shellfish poisoning toxins in Shellfish by ultra-performance rapid resolution LC-MS/MS. J AOAC Int 94:555–564
Turner A, Hatfield R (2012) Refinement of AOAC official method (SM) 2005.06 liquid chromatography-fluorescence detection method to improve performance characteristics for the determination of paralytic shellfish toxins in king and queen scallops. J AOAC Int 95:129–142
Halme M, Rapinoja M, Karjalainen M, Vanninen P (2012) Verification and quantification of saxitoxin from algal samples using fast and validated hydrophilic interaction liquid chromatography–tandem mass spectrometry method. J Chromatogr B 880:50–57
Campas M, Garibo D, Prieto-Simon B (2012) Novel nanobiotechnological concepts in electrochemical biosensors for the analysis of toxins. Analyst 137:1055–1067
Van Dolan F, Fire S, Leighfield T, Mikulski C, Doucette G (2012) Determination of paralytic shellfish toxins in shellfish by receptor binding assay: collaborative study. J AOAC Int 95:795–812
Vale C, Alfonso A, Vieytes M, Romarís X, Arévalo F, Botana A, Botana L (2008) In vitro and in vivo evaluation of paralytic shellfish poisoning toxin potency and the influence of the pH of extraction. Anal Chem 80:1770–1776
Schantz E, Ghazarossian V, Schnoes H, Strong F, Springer J, Pezzanite J, Clardy J (1975) Letter: the structure of saxitoxin. J Am Chem Soc 97:1238–1239
Humpage A, Magalhaes V, Froscio S (2010) Comparison of analytical tools and biological assays for detection of paralytic shellfish poisoning toxins. Anal Bioanal Chem 397:1655–1671
Henao-Escobar W, del Torno-de Román L, Domínguez-Renedo O, Alonso-Lomillo M, Arcos-Martínez M (2016) Dual enzymatic biosensor for simultaneous amperometric determination of histamine and putrescine. Food Chem 190:818–823
Sadik O, Yan F (2004) Novel fluorescent biosensor for pathogenic toxins using cyclic polypeptide conjugates. Chem Commun 7:1136–1137
Chen H, Kim Y, Keum S (2007) Surface plasmon spectroscopic detection of saxitoxin. Sensors 7:1216–1223
Vilariño N, Fonfría E, Louzao M, Botana L (2009) Use of biosensors as alternatives to current regulatory methods for marine biotoxins. Sensors 9:9414–9443
Tuerk C, Gold L (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249:505–510
Kirsch J, Siltanen C, Zhou Q, Revzin A, Simonian A (2013) Biosensor technology: recent advances in threat agent detection and medicine. Chem Soc Rev 42:8733–8768
Hayat A, Marty J (2014) Aptamer based electrochemical sensors for emerging environmental pollutants. Front Chem 2:41
Rhouati A, Yang C, Hayat A, Marty J (2013) Aptamers: a promising tool for ochratoxin a detection in food analysis. Toxins 5:1988–2008
Xu W, He J, Gao L, Zhang J, Yu C (2015) Immunoassay for netrin 1 via a glassy carbon electrode modified with multi-walled carbon nanotubes, thionine and gold nanoparticles. Microchim Acta 182:2115–2122
Moghaddam F, Taher M, Behzadi M, Naghizadeh M (2015) Modified carbon nanotubes as a sorbent for solid-phase extraction of gold, and its determination by graphite furnace atomic absorption spectrometry. Microchim Acta 182:2123–2129
Li S, Yan Y, Zhong L, Liu P, Sang Y, Cheng W, Ding S (2015) Electrochemical sandwich immunoassay for the peptide hormone prolactin using an electrode modified with graphene, single walled carbon nanotubes and antibody-coated gold nanoparticles. Microchim Acta 182:1917–1924
Su L, Gao F, Mao L (2006) Electrochemical properties of carbon nanotube (CNT) film electrodes prepared by controllable adsorption of CNTs onto an alkanethiol monolayer self-assembled on gold electrodes. Anal Chem 78:2651–2657
Handy S, Yakes B, DeGrasse J, Campbell K, Elliott C, Kanyuck K, DeGrasse S (2013) First report of the use of a saxitoxin-protein conjugate to develop a DNA aptamer to a small molecule toxin. Toxicon 61:30–37
Zheng X, Hu B, Gao S, Liu D, Sun M, Jiao B, Wang L (2015) A saxitoxin-binding aptamer with higher affinity and inhibitory activity optimized by rational site-directed mutagenesis and truncation. Toxicon 101:41–47
Yadav S, Kumar A, Pundir C (2011) Amperometric creatinine biosensor based on covalently coimmobilized enzymes onto carboxylated multiwalled carbon nanotubes/polyaniline composite film. Anal Biochem 419:277–283
Cunniff P (1995) Official methods of analysis of AOAC International, 16th edn. AOAC International, Arlington
Wang Y, Maspoch D, Zou S, Schatz G, Smalley R, Mirkin C (2006) Controlling the shape, orientation, and linkage of carbon nanotube features with nano affinity templates. PNAS 103:2026–2031
Guo K, Wang Y, Chen H, Ji J, Zhang S, Kong J, Liu B (2011) An aptamer–SWNT biosensor for sensitive detection of protein via mediated signal transduction. Electrochem Commun 13:707–710
Furtado C, Kim U, Gutierrez H, Pan L, Dickey E, Eklund P (2004) Debundling and dissolution of single-walled carbon nanotubes in amide solvents. J Am Chem Soc 126:6095–6105
Kele P, Orbulescu J, Gawley R, Leblanc R (2006) Spectroscopic detection of Saxitoxin: an alternative to mouse bioassay. Chem Commun 14:1494–1496
Wang Q, Fang J, Cao D, Li H, Su K, Hu N, Wang P (2015) An improved functional assay for rapid detection of marine toxins, saxitoxin and brevetoxin using a portable cardiomyocyte-based potential biosensor. Biosens Bioelectron 72:10–17
Acknowledgments
The authors are grateful for financial support from the National Natural Science Foundation (grant number: 21375019, 21377024), Special-funded Program on National Key Scientific Instruments and Equipment Development (grant number: 2011YQ150072) of China, and the Program for Changjiang Scholars and Innovative Research Team in University (grant number: IRT15R11). The authors have declared no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All experiments were performed in compliance with the relevant laws and institutional guidelines, andthe institutional committee has approved the experiments.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 376 kb)
Rights and permissions
About this article

Cite this article
Hou, L., Jiang, L., Song, Y. et al. Amperometric aptasensor for saxitoxin using a gold electrode modified with carbon nanotubes on a self-assembled monolayer, and methylene blue as an electrochemical indicator probe. Microchim Acta 183, 1971–1980 (2016). https://doi.org/10.1007/s00604-016-1836-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-016-1836-1