
Abstract
The authors show that carbon fiber bundles electrochemically modified with the conducting polymer poly(3,4-ethylenedioxythiophene) (PEDOT) is a viable sorbent for online solid-phase microextraction (SPME) of the sulfonamides (sulfadiazine, sulfadimidine and sulfamethoxazole) prior to their determination by HPLC. The fibers were packed in a tube loop made from polyether ether ketone (PEEK) that was coupled to the HPLC system for online SPME. Preconcentration factors can reach values of up to 300, and the limit of detection (at an S/N ration of 3) can be as low as 0.05 ng⋅mL−1. The method was applied to the analysis of the sulfonamides in spiked rat plasma with intra-day and inter-day RSDs of <3.33 and <4.57 %, and with recoveries in the range from 91.7 to 97.8 % in spiked plasma. The in-tube SPME was also applied to the determination of the 3 sulfonamides in rat plasma after oral administration (tablet powder) with high sensitivity. In addition to its efficient extraction, the PEEK tube based SPME has chemical and mechanical stability under even harsh conditions.

Electrochemically modified carbon fibers were packed into polyether ether ketone (PEEK) tube and connect with HPLC system. Good stability and extraction efficiency over 300 fold have been obtained. Application in plasma samples exhibits high extraction efficiency, good reproducibility and recoveries.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Conducting polymer is a kind of burgeoning material with good conductivity. A great deal of research work has been done since its discovery [1, 2] , especially after Hideki Shirakawa’s work was awarded the Nobel prize in chemistry [3]. In terms of structure, conducting polymer is chemically or electrochemically synthesized by monomers, among which conjugate π-bond formed. The monomer for polymerization is changeable, so the structure can be designed goal-directed for particular application. As a result, conducting polymer has been extensively applied in supercapacitors [4], fuel cells [5], biosensors [6, 7] and so on.
The application of electrochemical means for solid-phase microextraction (SPME) was reviewed by Collins [8]. The conducting polymer sorbents are with high uniformity of deposits, good control of the film thickness. Moreover, rational design of molecular recognition layer toward target analyte can be obtained between the layer and the analyte [9]. Application of conducting polymer in solid-phase microextraction [10, 11], such as polyaniline [12], polypyrrole [13], polythiophene [14] and their derivates [15–18] has already been reported. In these studies, conducting polymer is coated on a sort of substrate as sorbent. After extraction, the analytes was eluted by heating in gas chromatography or a volume of eluent. However, for thermo-labile compounds the extraction and elution step are inconsecutive, and the analytes may not be completely desorpted, leading to inaccuracy of extraction efficiency. It is essential to develop a universally incorporate extraction-detection method.
Recently Zhongze Gu’s group reported poly(3,4-ethylenedioxythiophene) (PEDOT) nanoclusters for solid phase extraction [19]. A small amount of PEDOT nanoclusters was employed as sorbent and placed in a pipette tip with glass wool on either side. Without complex preparation, 20 kinds of sulfonamides in honey were directly extracted from the sample. Although good extraction performance been achieved, there still exist some problems to overcome.
As a result, we employ carbon fiber bundle as substrate and PEDOT film is electrochemically coated on the surface of the fiber bundle. Carbon fiber is a kind of inorganic polymer fibers, with good thermal stability and electrical conductivity. Composing of carbon atoms like graphite endures it good stability in polar condition. A carbon fiber branch occupies large specific surface area. Its chemical inertness provides big room for surface modification to get desired structure. After modification the fiber bundle was packed into a polyether ether ketone (PEEK) tube. Then the tube loop was coupled to the HPLC system to set up an online SPME-HPLC system for extraction and detection of 3 sulfonamides as a model. PEDOT, a derivative of polythiophene conducting polymer, is composed of 3,4-ethylenedioxythiophene and can form nanoclusters and films. Consequently, we immobilized the PEDOT film onto the conductive substrate by electro polymerization and packed the coated substrate into a tube to establish an online SPME-HPLC system. Our method showed good selectivity and simplicity and ultra high extraction efficiency. We believe this strategy also can be applied to other target analytes in complex matrix by altering sorbent.
Experimental
Reagents and apparatus
Three sulfonamides standard and 3, 4-ethylene dioxythiophene (EDOT) monomer were purchased from Aladdin (http://www.sigmaaldrich.com). Unmodified bare carbon fibers were purchased from Shanghai Synthetic Fiber Research Institute (http://www.chemfiberinst.com). PEEK tubes (1/16 in. o.d, 0.02 in.i.d.) were obtained from Haohai Chemical (www.haohaigroup.com). Water purified by a Milli-Q system (≥18 MÙ · cm, http://www.emdmillipore.com/US/en) was used to prepare all solutions and mobile phases. Tablets of three sulfonamides were got from Shanxi Tianyuan Pharmaceutical Co., LTD (http://www.yaoxie.net.com). Sprague Dawley (SD) rats (about 250 g) were obtained from Animal Experimental Center, Wuhan University. Other reagents used were all of analytical grade.
An electrochemical analyzer CHI 842B was purchased from Chenhua Instrument Factory (Shanghai, China, http://www.chinstruments.com/) and parameters were set by electrochemical workstation. The HPLC system consists of two Shimadzu 20A pumps, a Shimadzu six-port valve (http://www.shimadzu.com.cn), a Shimadzu 20A degasser, 20A UV detector and thermostat controlled column compartment (Fig. 1). A syringe pump for sampling was from Baoding Longer Precision Pump Co., Ltd (Baoding, China, http://www.lgpump.com.cn). Data collection was performed on Shimadzu LC Solution software. Separation column was a C-18 column (150 mm × 4.6 mm i.d.) with 5 μm particle size from GL Science (Tokyo, Japan, http://www.glsciences.com). The mobile phase for isocratic elution consisted of methanol-water (containing 0.1 % formic acid) (43:57, v/v) and the flow rate was 0.7 mL⋅min−1. The detection wavelength was set at 269 nm and column temperature was 30 °C.

Schematic of online solid-phase microextraction-HPLC
The morphology images of PEDOT layer were obtained by using a field emission scanning microscope (Zeiss Ultra Plus, Germany, http://www.zeiss.com). Fourier transform infrared (FTIR) spectra were recorded by IR spectrometer (Thermo Nexus, USA, http://www.thermonexus.com).
Preparation of poly(3,4-ethylenedioxythiophene)-coated carbon fiber
The PEDOT layer was modified on carbon fiber by cyclic voltammetry carried out with electrochemical analyser CHI 842B. Before coating, the surfaces of the fiber bundle (0.0045 g, 12 cm) was cleaned with perchloric acid and rinsed with 0.1 mol⋅L−1 NaOH solution and double-distilled water and ultrasonicated in methanol. After removing inorganic impurities and oxide layer, one end of the bare fiber bundle (1 cm) was immobilized on the clamp of the three electrode system. The remaining part was immersed in supporting electrolyte solution used as work electrode. Platinum wire was used as counter electrode and saturated calomel as reference electrode. Perchloric acid containing EDOT monomer of certain concentration served as supporting electrolyte after removal of oxygen by sparging with nitrogen. Before modification, the fiber bundle was sonicated in methanol and water in turn and then applied potential between −0.2 ~ 0.8 V in potassium ferricyanide solution until symmetric redox peaks appeared. Voltage between −0.05 ~ +0.75 V was applied and proper scan rate and scan segments was chosen for the cyclic voltammetry accompanied with constant magnetic stirring (Fig. S1). The fiber bundle was washed and heated at 100 °C for 1 h then to immobilize the film. The polymerizing mechanism was shown in Fig. S2. After reaction, the modified fiber bundle was packed manually into blank PEEK tube (14 cm) and methanol was pushed through by a syringe pump to remove the residual reactants. The PEEK tube was dried in an oven (60 °C) for further use.
Stock solutions and samples
Stock solutions of 3 sulfonamides were prepared at the concentration of 50 mg⋅mL−1 in methanol. The sample solutions were prepared weekly by diluting standard mixture solution with the appropriate amount of water adjusted to a certain pH value with 0.1 mol⋅L−1 HCl and NaOH. The internal standard solution of 500 ng⋅mL−1 was prepared by diluting stock solution in methanol. A volume of diluted solution of internal solution was added to sample solutions to form a final concentration of 500 pg⋅mL−1. All solutions were stored at 4 °C before usage
Extraction procedure of solid-phase microextraction-HPLC
The PEEK tube was connected directly to the six-port valve of HPLC by two connectors (Fig. 1). Before extraction, a volume of methanol was replenished in by the syringe pump to remove impurities. Sample solution was made by diluting 20 μL sulfonamides-methanol solution to 20 mL with water. Online SPME–HPLC analysis was performed in two steps. In the sample loading step, the six-port valve was set to load mode while mobile phase was introduced directly into the column. Sample solution (20 mL) was introduced by a syringe pump and flowed through the PEEK tube into wastes bottle for SPME. Then in the eluting and separating step, the six-port valve was switched to injection mode and mobile phase flowed through the PEEK tube to elute the analytes into the HPLC column for separation and UV detection. Desorption process was realized by simply letting in the mobile phase flow through the PEEK tube. Because of the high strength of the PEEK tube and big interspace among the fibers, the fiber endures relatively high flow rates. Therefore, the desorption and HPLC separation process was reduced to a single step [20].
Solid-phase microextraction-HPLC analysis of sulfonamides in rat plasma
Blank rat plasma was obtained from healthy SD rats. For method validation, 0.3 mL spiked plasma was measured and put into a 1.5 mL eppendorf tube and then 0.6 mL methanol was added for deproteinization. Proteins were removed by centrifugation (624 g, 15 min) and the supernatant was collected, and dried under nitrogen. The residual was redissolved by 0.6 mL methanol and centrifuged (6932 g, 15 min) to get the supernatant. Purified water was added in 20 μL supernatant then to make a 20 mL solution, which was loaded onto the extraction system. The procedures and conditions were the same as mentioned above. For analyzing of sulfonamides concentration in plasma, 0.3 mg Tablets powder of three sulfonamides were dissolved in 1 mL purified water was administrated to a SD rat (250 g). Blood samples (5 mL) were obtained from the ophthalmic artery 1 h after a single oral administration and transferred to a heparinized Eppendorf tube and centrifuged (624 g, 15 min). 0.3 mL rat plasma was measured. After deproteinization, 20 mL sample was made for SPME–HPLC analysis.
Evaluation of extraction efficiency
The extraction procedure was performed by diluting the sample solution to 1000 fold and then extracted by the system. If the analytes were totally extracted on the sorbent and the peak area was equal to that before dilution (direct injection), the theoretical Extraction Efficiency (EE) would be 1000. Concerning about the concentration effect of SPME and possible loss of analytes in solution, the practical EF might be lower than theoretical result. Put it another way, it can be described in the following formula.
As in the formula was the peak area of standard analytes sample without extraction while Ae represented peak area of analytes with SPME. By keeping the total amount of analytes equal, standard sample solution (500 ng⋅mL−1) of 20 μL was directly injected into HPLC and the peak area (As) was obtained for evaluation of extraction efficiency.
Results and discussion
Optimization of polymerization conditions
The polymerization process of EDOT on carbon fiber may be strongly influenced by many factors, such as monomer and supporting analyte concentration, scan rates [21]. To obtain higher extraction efficiency, the following main affecting parameters were optimized: (a) Effect of monomer concentration; (b) scan rates; (c) scan segments. Respective data are given in the Electronic Supporting Material (Fig. S4). The following experimental conditions were found to give best results: (a) A monomer concentration of 5 mmol⋅L−1; (b) scan rates of 0.025 V⋅s−1; (c) 90 scan segments.
Characterization of modified carbon fiber bundles
During modification, the cyclic voltammogram was recorded simultaneously with the electronic polymerizing process (Fig. S1). The morphology of the modified fiber under optimal condition was examined by Scanning electron micrographs. As shown in Fig. 2, it was found that the materials were composed of small grains. These grains were with the size distribution from 100 to 250 nm. Considerable porosity existed in the membrane formed by the void spaces between the nano-grains domains. This structure provides large specific surface area for the extraction of the targets.

Scanning electron micrographs of modified carbon fiber: (a) and (b) bare carbon fiber;(c) and (d) modified carbon fiber
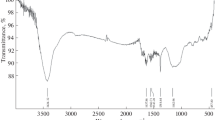
To verify the chemical composition of the PEDOT membranes, we investigated the principal vibrational modes of our materials by using FTIR spectra. As shown in Fig. S3, the spectra of PEDOT membranes appeared many peaks in the wave numbers range from 500 to 2000 cm−1. The peaks appearing at 1462 and 1384 cm−1 underpin the presence of C—C and C = C stretch vibrations of the thiophene ring [22]. Peaks at 983, 878, and 689 cm−1 are attributed to carbon-sulfur bonds in the thiophene ring [23], while peaks at 1191, 1099–1059 cm−1 were assigned to the alkylene-dioxy group [24]. These positions of the prominent bands appeared in finger print region matched with those reported earlier for polymerized PEDOT, indicating successful synthesis of the PEDOT membrane.
Stability of the modified carbon fiber bundle
The stability of PEDOT membrane is essential for application to SPME. As a result, the modified bundle was tested by treatment with harsh conditions including organic solvent, strong acidic, basic solutions and continuous usage.
The modified fiber bundles were first immersed in some commonly used organic solvents such as methanol, ethanol, isopropanol, dodecanol, acetonitrile, acetone, toluene, dimethylsulfoxide and chloroform with several replicates respectively. After being sealed and sonicated, the membranes were observed to be almost unchanged within 12 h, indicating good stability of the PEDOT membrane in organic solvents. The influence of acidic and basic conditions was also investigated by immersing the modified fiber in HCl and NaOH solutions of different concentrations (0.01, 0.1, 1 mol⋅L−1) separately. The layer was still stable even after being treated with 1 mol⋅L−1 HCl solution. However, when being treated with NaOH solutions with concentration above 0.1 mol⋅L−1, slight turbidity was observed, which was possibly due to decomposition of the PEDOT membrane under strong basic conditions. When being treated with NaOH solutions with concentration below 0.1 mol⋅L−1, the drop of PEDOT membrane was negligible. However,the influence of strong basic condition was circumvented, as most of SPME were performed under relatively moderate conditions.
Concerning about cost, the device should be recyclable. In the experiment we measured fifty times continuously. The 50th total peak area for three sulfonamides we got was 97.62 % of the first time measured, indicating good stability of the fiber after usage of many times.
Analysis of standard sample by online solid-phase microextraction-HPLC
The extraction efficiency and performances of the system were examined by analyzing three kinds of sulfonamides aqueous samples. The sorption of sulfonamides molecules onto the surface of modified fibers is mainly due to the hydrophobic interaction, hydrogen bonds and electrostatic force (Fig. 3). To obtain higher extraction efficiency, the following parameters that have potential effect on extraction efficiency were optimized: (a) pH value; (b) flow rate of pre-extraction solution; (c) volume of pre-extraction solution. Respective data are given in the Electronic Supporting Material (Fig. S5). The following experimental conditions were found to give best results: (a) pH 6; (b) 0.5 mL⋅min−1 of flow rate for pre-extraction solution; (c) 20 mL pre-extraction solution.

Hypothesis of interaction between poly(3,4-ethylenedioxythiophene) and sulfonamides
Extraction efficiency of the system
We evaluated the efficiency by the formula mentioned above. The extraction procedure was performed within 15 min under the optimized conditions. The extraction efficiency was calculated as about over 300 fold, suggesting that the PEDOT-electrochemically modified carbon fiber showed exceptional selectivity toward sulfonamides. As shown in Fig. 4, the chromatogram of standard sample (500 ng⋅mL−1) as well as the chromatogram after extraction (standard sample diluted to 1000 fold at concentration of 0.5 ng⋅mL−1) indicates good selectivity of the sorbent toward sulfonamides.

Chromatograms of standard aqueous solution: a Direct injection of 500 ng⋅mL−1; b extraction after dilution of 1000 fold. 1 sulfadiazine, 2 sulfadimidine and 3 sulfamethoxazole
Analytical performance for standard samples
The low limits of detection (LODs) and the repeatability were performed for analysis of three sulfonamides in aqueous samples under optimized conditions (Table 1). The obtained linearity was in the range of 0.05–15 ng⋅mL−1 with R higher than 0.9993. LODs (S/N = 3) were found to be as low as 0.05 ng⋅mL−1, allowing trace determination of sulfonamides in aqueous samples. By loading six replicates of standards sample solution (0.5 ng⋅mL−1), RSDs for peak areas were calculated and found to be lower than 5 %, indicating good reproducibility of the method.
Comparison of the method with reported methods
Sensitivity is crucial for a method as to determination of trace amount of analytes. Therefore, the method was also compared with other SPME methods reported for sulfonamides extraction and determination [19, 25–28], mainly concerning sensitivity. As shown in Table 2, the LOD of the method was lower than most of reported SPME method, and even better than MS/MS detection. The results indicate the superiority of the PEDOT-based online SPME-HPLC method.
Application in analysis of rat plasma samples
The extraction procedure for rat plasma sample was similar to that of standard samples. The plasma sample was made of plasma (0.3 mL),sample solution (0.3 mL) and methanol (0.6 mL) . From the chromatograms of spiked plasma samples showed in Fig. 5a, we can see the peaks for matrix mainly emerged within 2 to 5 min. After SPME, sulfonamides were well extracted and most of the matrices are removed (Fig. 5b). Analytical performances of sulfamethoxazole in rat plasma sample were investigated, and parameters including LOD, correlation coefficients, and the repeatability were evaluated under the optimized conditions (Table 3). Good linearity was obtained in the range of 0.5–50 ng⋅mL−1 in plasma sample. As compared with the method in standard solutions, the sensitivity was reduced in plasma sample analysis. However, LOD and LOQ measured were sufficiently sensitive for plasma concentration monitoring [29]. The calculated RSDs for intra-day and inter-day precession were found to be lower than 3.9 %, exhibiting good reproducibility of the method in plasma sample analysis (Table. S1).

Chromatograms of spiked plasma samples: A Direct injection, B with online solid-phase microextraction-HPLC, and C plasma sample from SD rat after oral administration of sulfonamides tablets powder. 1 sulfadiazine, 2 sulfadimidine and 3 sulfamethoxazole
Recoveries were also investigated in the spiked rat plasma samples (Table S2). Blank plasma was spiked with three sulfonamides standards and diluted to concentrations of 0.5, 1.5 and 5 ng⋅mL−1, respectively. The amounts of recovered sulfonamides were determined and the calculated recoveries obtained were in the range of 91.7–97.8 %, which showed good accuracy of this method.
Monitoring drug concentration of rat plasma after oral administration of sulfonamides tablets powder
The method was further applied to monitor drug concentration in rat plasma after oral administration of three sulfonamides tablets powder, which are widely used in agriculture as anti-microbial, anti-plasmodial drug. 1 mL tablets powder aqueous suspension of 0.1 mg⋅mL−1 was given to a SD rat and the concentrations in plasma were determined. A typical chromatogram is shown in Fig. 5c. As can be seen in the chromatogram, the peaks for sulfonamides are well identified and separated with other components of tablets and the plasma matrix. The drug concentration in plasma was calculated to be 0.8, 0.73 and 0.57 ng⋅mL−1 for sulfadiazine, sulfadimidine and sulfamethoxazole, respectively. The results indicate that this method can be used for monitoring concentration of three sulfonamides in plasma.
Conclusion
We report a feasible strategy of in-tube SPME–HPLC based on conducting polymer coated carbon fiber bundles. The fiber bundles were packed inside PEEK tube loop for sulfonamides analysis in aqueous and plasma samples. The coated fibers were stable under harsh conditions. Good stability and extraction efficiency over 300 fold showed with the method. Application in plasma samples exhibits high extraction efficiency, good reproducibility and recoveries. We can conclude that the electrochemical modification method is reliable to immobilize conducting polymer for high selective extraction of target molecules. The method can be used for online SPME-HPLC analysis of sulfonamides in plasma samples. The system would be effective for the analysis of other analytes in aqueous samples by simply changing the composition of the polymer sorbents.
References
Huang WS, Humphrey BD, MacDiarmid AG (1986) Polyaniline, a novel conducting polymer. Morphology and chemistry of its oxidation and reduction in aqueous electrolytes. J Chem Soc Faraday Trans 82:2385
Huang JC (2002) Carbon black filled conducting polymers and polymer blends. Adv Polym Technol 21:299
Shirakawa H (2001) The discovery of polyacetylene film: the dawning of an era of conducting polymers (Nobel lecture). Angew Chem-Int Ed 40:2574
Snook GA, Kao P, Best AS (2011) Conducting-polymer-based supercapacitor devices and electrodes. J Power Sources 196:1
Kreuer KD (2001) On the development of proton conducting polymer membranes for hydrogen and methanol fuel cells. J Membr Sci 185:29
Gerard M, Chaubey A, Malhotra BD (2002) Application of conducting polymers to biosensors. Biosens Bioelectron 17:345
Xia L, Wei Z, Wan M (2010) Conducting polymer nanostructures and their application in biosensors. J Colloid Interface Sci 341:1
Collins CJ, Arrigan DW (2009) A review of recent advances in electrochemically modulated extraction methods. Anal Bioanal Chem 393:835
Abdolmohammad-Zadeh H, Rezvani Z, Sadeghi GH et al (2011) Layered double hydroxides: a novel nano-sorbent for solid-phase extraction. Anal Chim Acta 685:212
Wei F, Fan J, Zheng MM et al (2010) Combining poly (methacrylic acid‐co‐ethylene glycol dimethacrylate) monolith microextraction and octadecyl phosphonic acid‐modified zirconia‐coated CEC with field‐enhanced sample injection for analysis of antidepressants in human plasma and urine. Electrophoresis 31:714
Ma Q, Chen M, Shi ZG et al (2009) Preparation of a poly (N‐isopropylacrylamide‐co‐ethylene dimethacrylate) monolithic capillary and its application for in‐tube solid‐phase microextrac‐tion coupled to high‐performance liquid chromatography. J Sep Sci 32:2592
Li X, Zhong M, Xu S et al (2006) Determination of phthalates in water samples using polyaniline-based solid-phase microextraction coupled with gas chromatography. J Chromatogr A 1135:101
Wu J, Pawliszyn J (2004) Solid-phase microextraction based on polypyrrole films with different counter ions. Anal Chim Acta 520:257
Li X, Li C, Chen J et al (2008) Polythiophene as a novel fiber coating for solid-phase microextraction. J Chromatogr A 1198:7
Banitaba MH, Mohammadi AA, Davarani SSH et al (2011) Preparation and evaluation of a novel solid-phase microextraction fiber based on poly (3, 4-ethylenedioxythiophene) for the analysis of OCPs in water. Anal Methods 3:2061
Wu M, Zhang H, Zhao F et al (2014) A novel poly (3, 4-ethylenedioxythiophene)-ionic liquid composite coating for the headspace solid-phase microextraction and gas chromatography determination of several alcohols in soft drinks. Anal Chim Acta 850:41
Ma Y, Zhao F, Zeng B (2013) Electrodeposition of poly (3, 4-ethylenedioxythiophene) on a stainless steel wire for solid phase microextraction and GC determination of some esters with high boiling points. Talanta 104:27
Hosseiny Davarani SS, Nojavan S, Asadi R et al (2013) Electro‐assisted solid‐phase microextraction based on poly (3, 4‐ethylenedioxythiophen) combined with GC for the quantification of tricyclic antidepressants. J Sep Sci 36:2315
Tian T, Zheng S, Ye B et al (2013) Poly-3, 4-ethylenedioxythiophene nanoclusters for high effective solid phase extraction. J Chromatogr A 1275:17
Yang N, Zoski CG (2006) Polymer films on electrodes: investigation of ion transport at poly (3, 4-ethylenedioxythiophene) films by scanning electrochemical microscopy. Langmuir 22:10338
Simonet J, Rault-Berthelot J (1991) Electrochemistry: a technique to form, to modify and to characterize organic conducting polymers. Prog Sol State Chem 21:1
Hernandez V, Ramirez FJ, Otero TF et al (1994) An interpretation of the vibrational spectra of insulating and electrically conducting poly (3‐methylthiophene) aided by a theoretical dynamical model. J Chem Phys 100:114
Martin DC, Wu J, Shaw CM et al (2010) The morphology of poly (3, 4-ethylenedioxythiophene). Polym Rev 50:340
Van Laar B, Ijdo DJW (1971) Preparation, crystal structure, and magnetic structure of LiCrS< sub> 2</sub> and LiVS< sub> 2</sub> J Solid State Chem 3:590
Tao Y, Liu JF, Hu XL et al (2009) Hollow fiber supported ionic liquid membrane microextraction for determination of sulfonamides in environmental water samples by high-performance liquid chromatography. J Chromatogr A 1216:6259
Lu KH, Chen CY, Lee MR (2007) Trace determination of sulfonamides residues in meat with a combination of solid-phase microextraction and liquid chromatography–mass spectrometry. Talanta 72:1082
Yu C, Hu B (2012) C 18-coated stir bar sorptive extraction combined with high performance liquid chromatography–electrospray tandem mass spectrometry for the analysis of sulfonamides in milk and milk powder. Talanta 90:77
Huang X, Qiu N, Yuan D (2009) Simple and sensitive monitoring of sulfonamide veterinary residues in milk by stir bar sorptive extraction based on monolithic material and high performance liquid chromatography analysis. J Chromatogr A 1216:8240
Deng Y, Liao Q, Li S et al (2008) Simultaneous determination of berberine, palmatine and jatrorrhizine by liquid chromatography–tandem mass spectrometry in rat plasma and its application in a pharmacokinetic study after oral administration of coptis–evodia herb couple. J Chromatogr B 863:195
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant nos. 81573384, 21375101 and 91417301), and Natural Science Foundation of Hubei (No. 2014CFA077).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 553 kb)
Rights and permissions
About this article

Cite this article
Ling, X., Zhang, W. & Chen, Z. Electrochemically modified carbon fiber bundles as selective sorbent for online solid-phase microextraction of sulfonamides. Microchim Acta 183, 813–820 (2016). https://doi.org/10.1007/s00604-015-1726-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-015-1726-y