
Abstract
This article describes an aptamer-based thrombin assay using a hemin-based peroxidase mimetic for signal amplification. Thrombin is recognized by an immobilized primary aptamer (G1-quadruplex). The G1-quadruplex/hemin complex formed quenches the fluorescence of CdTe quantum dots (QDs) due to photoinduced electron transfer (PET). In the next step, thrombin is associated with a secondary aptamer (G2-quadruplex) consisting of Pt nanoparticles (NPs), G2-quadruplex and hemin to form a sandwich structure. Both the G1-quadruplex/hemin complex and the Pt NPs/G2-quadruplex/hemin complex associated with thrombin act as enzyme mimetics and catalyze the oxidation of hydroquinone by H2O2 to form 2-hydroxy-p-benzoquinone (HPB). The HPB produced quenches the fluorescence of the CdTe QDs via a PET and an inner filter effect, thus causing large signal amplification. The effects were exploited to design a highly sensitive and selective thrombin assay. Under optimized conditions, a linear fluorescence response is achieved in the 0.05 pmol·L−1 to 10 nmol·L−1 concentration range, with a lower detection limit of 15 fmol·L−1. This approach, in our perception, represents a promising platform for sensitive detection of biomolecules for which appropriate aptamers can be found.

A sensitive fluorescent method was developed for the specific detection of thrombin. It is based on the photoinduced electron transfer (PET) induced fluorescence quenching of CdTe quantum dots (QDs) using enzyme mimetics for signal amplification.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Thrombin is a serine protease that plays a key function in hemostasis and blood clotting, which shows promotion for thrombosis, angiogenesis and inflammation [1]. Generally, the normal concentration of thrombin in blood ranges from nM to low μM during the coagulation process [2]. Therefore, it is very important to establish sensitive methods to detect thrombin for disease diagnosis and drug discovery.
Aptamers are a kind of protein-affinity tag that can bind to target molecules such as proteins with equal affinity to that of antibody [3]. Especially, aptamers reveal some evident advantages over antibodies. For instance, great resistance against denaturation, produced synthetically eliminating batch-to-batch variation, and simple chemical modification. Till now, many different techniques based on the thrombin binding aptamer (TBA) with high specificity and affinity toward thrombin have been developed for thrombin detection, such as electrochemistry [4], spectrophotometry [5, 6], chemiluminescence [7], surface enhanced Raman scattering (SERS) spectroscopy [8]. Among these methods, fluorescence assay, because of its distinct merits of inherent simplicity, high sensitivity, and short detection time, is attracting more and more research interest [9]. Notably, compared with traditional molecular fluorophores, as fluorescent probes, quantum dots (QDs) provide many potential advantages, such as size-tunable emission wavelength, superior brightness, and remarkable photostability. So, QDs hold good promise for ultrasensitive bioanalysis [10, 11]. It is demonstrated that energy transfer exists between QDs and complementary partners (acceptors or donors) upon excitation, which leads to photoluminescence (PL) change in QDs. Till now, QDs assisted thrombin sensing are mainly based on this fluorescence resonance energy-transfer (FRET) strategy [12, 13]. However, because the generation of FRET requires a strict distance between the donor and acceptor, it is inevitable to modify the QDs probe and/or biomolecules with tough and time-consuming integrated work. Thus, the design of convenient, rapid and inexpensive approaches for the sensitive and selective detection of thrombin is still demanding.
In contrast to the widely used FRET system for protein detection, aptamer sensing systems based on photoinduced electron transfer (PET) mechanism is scarce [14], although PET has been testified to be an effective approach to alter QDs’ fluorescence [15]. On the other side, natural enzymes or enzyme mimetics based signal amplification is attractive to achieve high sensitivity of the electrochemical [16], colorimetric [17] or chemiluminescent [18] aptamer-based assays. However, fluorescent aptamer-based assays with amplified signal production based on natural enzymes or enzyme mimetics are limited. Considering these aspects, in this work, a highly sensitive fluorescence aptamer-based assay was designed for thrombin assay. It is known that the TBA possesses a stable G-quadruplex structure after binding to thrombin, and a G-quadruplex/hemin structure was formed through intercalating hemin into TBA. This G1-quadruplex/hemin formed by the association of the primary aptamer with thrombin quenched the fluorescence of CdTe QDs due to PET from CdTe QDs to the intercalated hemin. Subsequently, Pt nanoparticles (NPs)/G2-quadruplex/hemin as a secondary aptamer was further conjugated to thrombin, forming a sandwich structure. The Pt NPs/G2-quadruplex/hemin as well as the G1-quadruplex/hemin formed by the association of the primary aptamer with thrombin exhibited fantastic catalytic ability for the oxidation of hydroquinone (HQ) to 2-hydroxy-p-benzoquinone (HPB). The mimicking enzymatically generated HPB readily interacted with CdTe QDs and quenched the fluorescence of QDs via PET and inner-filter effect, thus greatly amplified the signal readout. Due to the above multiple effects, this method was highly sensitive for thrombin with limit of detection (LOD) down to 15 fmol·L−1, which is relatively much lower than most other existing fluorescence assays for thrombin.
Experimental section
Reagents
Tellurium power, glutathione (GSH), CdCl2·2.5H2O, NaBH4, o-phenylenediamine (OPD), 3,3′-diaminobenzidine (DAB), hydroquinone (HQ), H2PtCl6·6H2O, N-hydroxysulfosuccinimide (NHS), tris (2-carboxyethyl) phosphine hydrochloride (TCEP), hemin (III) chloride, sodium citrate (Cit), H2O2 (30 wt%), L-tyrosine (L-Tyr), bovine albumin (BSA), monoethanolamine (MEA), lysozyme (LZM), L-Cysteine (L-Cys), tris-hydroxymethylaminomethane hydrochloride (tris), were all purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China, http://www.sinoreagent.com/). N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDC), poly (diallyldimethylammonium chloride) (PDDA) solution (20 %) were purchased from Sigma-Aldrich (St. Louis, USA, http://www.sigmaaldrich.com). Thrombin (enzymatic activity of 40–300 NIH units/mg, lyophilized power) was obtained from Shanghai Yuanye Biological Technology Co., Ltd. (Shanghai, China, http://www.shyuanye.com/); Human IgG was obtained from Wuhan Boster Biological Technology Co., Ltd. (Wuhan, China, http://www.boster.com.cn/). Thiol-terminated thrombin binding aptamer (TBA): 5′-SH-(CH2)6-GGT TGG TGT GGT TGG-3′; Amino-terminated thrombin binding aptamer (TBA’): 5′-NH2-(CH2)6- AGT CCG TGG TAG GGC AGG TTG GGG TGA CT-3′ were obtained from Sangon Biotech Co., Ltd. (Shanghai, China, http://www.sangon.com/). 20 mmol·L-1 tris-HCl buffer (pH 7.4) containing 140 mmol·L-1 NaCl, 5 mmol·L-1 KCl, 1 mmol·L-1 CaCl2 and MgCl2 were used as a buffer solution. 96-well plates (tissue culture treated, flat, clear bottom, black walls) were from Corning, Inc. (Corning, NY, http://www.corning.com/). A conventional dialysis bag was used to remove unreacted impurities, which obtained from Shanghai yuanye Bio-Technology Co., Ltd. with cut-off 8000–14,000.
Apparatus
Fluorescence measurements were performed on a SpectraMax M5 Multi-Mode Microplate Readers (Molecular Devices, USA). UV/vis absorption spectra were recorded by a TU-1901 spectrophotometer (Beijing Purkinje General Instrument Co., Ltd., China). Lifetime measurements were performed with a FLS920 photoncounting spectrometer (Edinburgh Instruments, UK). High resolution transmission electron microscopy (HRTEM) images were obtained on a JEOL JEM-2100 transmission electron microscope (Hitachi, Japan).
Synthesis of GSH-capped CdTe QDs
The preparation of GSH-capped CdTe QDs was performed according to literature [19]. Briefly, 12.8 mg Te powder and 11.3 mg NaBH4 were dissolved in 4 mL of ultrapure water (18.2 MΩ⋅cm) with ultrasound under N2 atmosphere to prepare fresh NaHTe. 91.3 mg CdCl2·2.5H2O and 0.8 mmol GSH was dissolved in 100 mL water, followed by adjusting the solution pH to 8.5–9.0 by dropwise addition of 1.0 mol·L−1 NaOH. Then 3.5 mL of the freshly prepared NaHTe solution was added and the resulting mixture was subjected to refluxing at 100 °C for 30 min. The prepared CdTe QDs were stored under dark before use.
Synthesis of Pt NPs
Pt NPs were prepared using a modified procedure [20]. Typically, 3 mL of 3.3 mmol·L−1 citrate (Cit) and 12 mL of 0.33 mmol·L−1 H2PtCl6 were mixed, followed by dropwise addition of 4 mL of 2.7 mmol·L−1 NaBH4 under stirring, and the mixture was allowed to react for 12 h at room temperature and then dialyzed for 1 day to remove impurities.
Preparation of the Pt NPs/G2-quadruplex/hemin complex
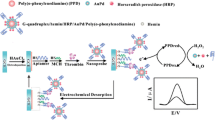
The fabrication process of the Pt NPs/G2-quadruplex/hemin complex is shown in Scheme 1A. Typically, 10 μL of thiol-terminated TBA (a secondary aptamer, 100 μmol·L−1) and 10 μL of 0.01 mol·L−1 TCEP were mixed for 2 h. Then, 980 μL of Pt NPs was added with an extra incubation time of 24 h. Subsequently, 300 μL of 20 mmol·L−1 tris-HCl buffers and 300 μL of 5.0 μmol·L−1 hemin were added into the mixture and reacted for 6 h under stirring. 200 μL BSA (w/w, 1 %) was implemented with a reaction time of 40 min. The final solution was then subjected to centrifuge at 8385 g for 20 min and then resuspended in 1.6 mL of tris-HCl buffer for further use.
Fabrication of the fluorescent aptamer-based assay
The protocol of the fluorescent aptamer-based assay for thrombin is illustrated in Scheme 1B. Firstly, 100 μL of 2 % PDDA was added into 96-well plates and incubated for 1 h. After washing by water for three times and drying by N2 flow, 80 μL of the negatively-charged CdTe QDs was added and incubated for 12 h, followed by washing three times with water. Then, 10 mg·mL−1 EDC and 5 mg·mL−1 NHS were dropped into the plates (50 μL·well−1) and incubated for 1 h in order to activated carboxyl of the GSH-capped CdTe QDs, followed by washing with tris-HCl. Subsequently, 30 μL of the amino-terminated thrombin binding aptamer (a primary aptamer, 0.5 μmol·L−1) solution was added into the plates and incubated for 16 h at room temperature, followed by washing with tris-HCl and blocking by 1 mmol·L−1 monoethanolamine (MEA) for 40 min. The fluorescence intensity of the CdTe QDs after the primary aptamer’ immobilization was recorded as F0. After that, 30 μL of different concentrations of thrombin was added into the plates and incubated for 40 min and then incubated with 30 μL of 5 μmol·L−1 hemin for 30 min. Finally, 30 μL of the Pt NPs/G2-quadruplex/hemin was put into the plates and incubated for another 30 min to form the sandwich aptamer-based assay. The wells were washed with tris-HCl before the addition of disparate solutions.
After the assembly of the “sandwich” fluorescence aptamer-based assay, 50 μL of 1.0 × 10−2 mol·L−1 hydroquinone (HQ), 50 μL of 1.0 × 10−3 mol·L−1 H2O2 and 100 μL of 20 mmol·L−1 tris-HCl (pH 7.4) were added into the plates and reacted for 5 min, the fluorescence intensity (designated as F) was measured with the excitation wavelength of 360 nm.
Results and discussion
Characterization of GSH-capped CdTe QDs, Pt NPs and Pt NPs/G2-quadruplex/hemin complex
CdTe QDs were chosen as the probe because they exhibit interesting properties such as large excitation Bohr radius, narrow emission band, and high photoluminescence [21]. As shown in Fig. 1, the CdTe QDs’ aqueous solution had a clear UV-vis absorption peak at 505 nm and a strong fluorescent emission peak at 537 nm. The CdTe QDs had a narrow fluorescence spectrum and the full-width at half-maximun was about 40 nm, meaning that they were relatively uniform in size. The average size of the CdTe QDs was about 2.5 nm calculated by the equation D = (9.8127 × 10−7)λ3-(1.7147 × 10−3)λ2 + (1.0064)λ-(194.84) (D (nm) was the size of a given nanocrystal sample, and λ (nm) was the wavelength of the first excitonic absorption peak of the corresponding sample) [22]. The morphology and size information of the CdTe QDs were further confirmed by the TEM image (about 2–3 nm with standard deviation of 0.18, Fig. S1A). The as prepared Pt NPs had a spherical shape with diameters about 2–5 nm with standard deviation of 0.46 (Fig. S1B).

The UV-vis absorption (black, left axis) and the fluorescence spectra (blue, right axis) of the CdTe QDs
Thrombin binding aptamer (TBA) can form a stable G-quadruplex structure, as a consequence, the G-quadruplex/hemin structure is formed with the hemin intercalated into TBA [23, 24]. In this experiment, the combination of thiol-terminated TBA and Pt NPs was realized through a Pt-S bond. Thereafter, hemin was intercalated into TBA and finally yielded the Pt NPs/G2-quadruplex/hemin complex, which was used as identifying and signal magnifying components for thrombin. We used UV-vis absorption spectroscopy to confirm the formation of the Pt NPs/G2-quadruplex/hemin complex. As shown in Fig.S2, the Pt NPs did not have any characteristic absorption maximum (curve a). After the TBA molecules were attached onto the surface of Pt NPs, a 269 nm absorption peak corresponding to the typical DNA absorption appeared (curve b), indicating the successful loading of TBA on Pt NPs. Compared with the absorption spectrum of Pt NPs/TBA, a new absorption peak at approximately 396 nm appeared for the Pt NPs/G2-quadruplex/hemin (curve c). This absorption peak was close to that of free hemin (394 nm) in solution (Fig. S2, inset), meaning that hemin was intercalated on the Pt NPs/TBA through the formation of G-quadruplex. Pt NPs, due to its relatively large surface area with multiple active sites, can catalyze the oxidation of many substrates, such as 3,3′,5,5′-tetramethylbenzidine (TMB) [25], uric acid [26], etc., using H2O2 as an oxidant, whose function is identical to that of the natural peroxidase, We chose Pt NPs but not Ag or Au ones because we found that Pt NPs showed better peroxidase-like activity (data not shown). Hemin can embed into TBA to form a G-quadruplex/hemin structure which also exhibits higher peroxidase-like catalytic ability than free hemin [16]. In this work, we found that not only the Pt NPs but also the G-quadruplex/hemin catalyzed the oxidation of HQ to HPB. The HPB had an orange colour and a characteristic absorption peak at around 480 nm (Fig. 2) [27]. Compared with Pt NPs and the G-quadruplex/hemin, the formed Pt NPs/G2-quadruplex/hemin showed higher enzyme-like activity for the catalytic oxidation of HQ (Fig. 2). As indicated in Fig.S3, when HQ was oxidized to HPB by H2O2 using Pt NPs/G2-quadruplex/hemin and G1-quadruplex/hemin as catalytic signal amplifiers, the fluorescence of CdTe QDs quenched obviously, while HQ (2.5 mmol·L−1) or H2O2 (0.25 mmol·L−1) alone or their mixture hardly affected the fluorescence intensity of CdTe QDs (Fig.S3). So, we inferred that the oxidation product of HQ (herein HPB) catalyzed by Pt NPs/G2-quadruplex/hemin and G1-quadruplex/hemin was an efficient quencher for the fluorescence of CdTe QDs.

UV-vis spectra of the oxidation of HQ catalyzed by (a) Pt NPs/G2-quadruplex/hemin, (b) Pt NPs, and (c) G2-quadruplex/hemin. Inset shows the corresponding color change of the catalytic systems
Fluorescence detection of thrombin with the aptamer-based assay
The protocol of the newly fluorescent aptamer-based assay for thrombin detection is illustrated in Scheme 1. This involves fixation of CdTe QDs on a 96-well plate, covalent conjugation of the amino-terminated primary TBA on the CdTe QDs, association of the thrombin target with the primary TBA and subsequent hemin intercalation to form G1-quadruplex/hemin conjugated thrombin. Considering that one thrombin molecule has two binding domains for its aptamers [16, 28–30], the as prepared Pt NPs/G2-quadruplex/hemin as a secondary aptamer associated with thrombin, forming a “sandwich” structure.

Illustration of the preparation of the fluorescent aptamer-based assay and the detection mechanism
As was observed, fluorescence quenching occurred after the primary aptamer’s association with thrombin and intercalating hemin (Fig. S4). We ascribed this fluorescence quenching to PET between CdTe QDs and hemin intercalated in the G1-quadruplex structure [14]. CdTe QDs had a conduction band (CB) edge at −1.0 V vs normal hydrogen electrode (NHE) [31], while the hemin(III) in the G1-quadruplex/hemin complex exhibited a reduction potential around −0.161 V vs NHE [32] corresponding to the FeIII/FeII-protoporphyrin IX couple. A comparison of the reduction potential of the G1-quadruplex/hemin complex and the CB edge of the CdTe QDs (Scheme 1C) implied that the excited electrons of the CdTe QDs transferred to the nearby hemin complex thermodynamically.
When more thrombin was present, more Pt NPs/G2-quadruplex/hemin was expected to be captured and more G1-quadruplex/hemin was formed, both of which catalyzed the oxidation of HQ to generate HPB. As discussed previously, HPB was an efficient quencher for CdTe QDs. So, the catalytic reaction by the formed biocatalytic structures (Pt NPs/G2-quadruplex/hemin complex and G1-quadruplex/hemin) further amplified fluorescent signal output (Fig. S4). In order to clarify the fluorescence quenching mechanism of CdTe QDs by the HPB, lifetime measurement was made to identify dynamic or static quenching [33]. Dynamic quenching refers to a process that involves the fluorophore and the quencher coming into contact during the transient existence of the excited state. Static quenching means the formation of fluorophore-quencher complex. The lifetime of CdTe QDs was found to decay obviously in the presence of HPB (Fig. 3), suggesting that it was a dynamic quenching. The fluorescence decay spectra were fitted with two decay components, which gave a major component in 16.2 ns, 96.59 % for free CdTe QDs, and 0.45 ns, 61.30 % in the presence of HPB (Table S1). Thus, we inferred that the CdTe QDs showed an obviously shorter fluorescence lifetime in the presence of HPB, presumably due to PET from CdTe QDs to HPB (Scheme 1B). As was observed by us and other groups, molecules with benzoquinone groups can act as an efficient electron acceptor of QDs [34]. In addition, the strong absorption of HPB at 360 nm (i.e. the exCitation wavelength for QDs) caused inner filter effect (IFE). Based on the above analysis, a conclusion can be made that HPB quenched the fluorescence of QDs through PET and IFE.

Fluorescence decay time of CdTe QDs in the absence (black line) and presence (red line) of HPB obtained by enzyme mimetics catalyzed oxidation of HQ
To obtain a high sensitivity, we optimized the experimental conditions such as substrates of the enzyme mimetics, the pH of the catalytic solution, incubation time between the thrombin and the aptamer, and the catalytic reaction time for producing HPB. Results shows that hydroquinone (HQ) as the substrate demonstrates the best quenching efficiency than that of o-phenylenediamine (OPD), 3,3ˊ-diaminobenzidine (DAB), or L-tyrosine (L-Tyr) (Fig. S5). The optimum solution pH for the catalytic reaction was found at 7.4 (Fig.S6) and the incubation time between thrombin and the primary TBA or the secondary aptamer tended to reach constant after 40 and 30 min, respectively (Fig.S7 A and B). The catalytic reaction for producing HPB was completed after 6 min (Fig.S7C).
Under the optimum experimental conditions, we explored the quantitative range of the FL aptamer-based assay. As shown in Fig.4, the FL intensity decreased progressively with the increase of the concentration of thrombin and a good linear relationship was found between the FL quenching effect (F0/F, where F0 and F are the FL intensity of the system in the absence and presence of thrombin, respectively) and the concentration of thrombin in the range of 0.05 pmol·L−1 to 10 nmol·L−1. The regression equation was F0/F = 1.78 + 0.42 Log C (pmol·L−1) and a detection limit of 15 fmol·L−1 was obtained (estimated according to S/N = 3). The in situ generated G1-quadruplex/hemin as well as the large quantity of HPB produced by the biocatalysts of the Pt NPs/G2-quadrupex/hemin and G1-quadrupex/hemin, resulting in multiple signal amplifying strategy. These multiple amplified PET protocol made the strategy among the most sensitive approach for aptamer-based thrombin monitoring (Table S2). The detection limit of the method was much lower than most other methods. Furthermore, other non-target molecules such as BSA, LZM, human IgG and L-Cys were tested to evaluate the selectivity of the aptamer-based assay (Fig.5). We can see that the presence of the interference molecules had little influence on the fluorescence intensity of the biosensor, revealing the good selectivity of the approach.

a Normalized FL spectra of the aptamer-based assay for thrombin detection at different concentrations: 0 pmol·L−1, 0.05 pmol·L−1, 0.1 pmol·L−1, 5 pmol·L−1, 0.01 nmol·L−1, 10 nmol·L−1. b The relationship between quenching effects (F0/F) and the concentration of thrombin. Detection buffer: tris-HCl (pH = 7.4) containing 2.5 mmol·L−1 HQ and 0.25 mmol·L−1 H2O2

Selectivity of the aptamer-based assay for the detection of thrombin (0.1 nmol·L−1) against the interference molecules, BSA (100 nmol·L−1), LZM (100 nmol·L−1), human IgG (100 nmol·L−1) and L-Cys (100 nmol·L−1). The error bar indicates the relative standard deviation of four repeated experiments
Conclusions
In this work, we demonstrated a highly sensitive fluorescent method for the detection of thrombin. The G1-quadruplex/hemin, thrombin and Pt NPs/G2-quadruplex/hemin were associated on the surface of CdTe QDs, forming a sandwich structure. Multiple fluorescence quenching for the CdTe QDs occurred in the detection system: 1) PET from CdTe QDs to the hemin intercalated in the G1-quadruplex formed by the association of the primary aptamer with thrombin made a contribution to the signal readout; 2) with Pt NPs/G2-quadruplex/hemin and G1-quadruplex/hemin acting as enzyme mimetics for signal amplification, the catalytically generated oxidized product of HQ (that is, HPB) further quenched the fluorescence of CdTe QDs due to PET and IFE. Due to the multiple quenching effects, especially the enzyme mimetics based signal amplification, this immunoassay possessed higher sensitivity and wider linear range. The protocol holds a promising perspective for providing a highly sensitive method for the biological detection.
References
Davie EW, Fujikawa K, Kisiel W (1991) The coagulation cascade: initiation, maintenance, and regulation. Biochem 30:10363–10370
Sun AL, Jia FC, Zhang YF, Wang XN (2014) Gold nanocluster-encapsulated glucoamylase as a biolabel for sensitive detection of thrombin with glucometer readout. Microchim Acta 182:1169–1175
Ellington AD, Szostak JW (1990) In vitro selection of RNA molecules that bind specific ligands. Nat 346:818–822
Zhang HF, Shuang SM, Sun LL, Chen AJ, Qin Y, Dong C (2014) Label-free aptasensor for thrombin using a glassy carbon electrode modified with a graphene-porphyrin composite. Microchim Acta 181:189–196
Xu ZC, Huang XY, Dong CQ, Ren JC (2014) Fluorescence correlation spectroscopy of gold nanoparticles, and its application to an aptamer-based homogeneous thrombin assay. Microchim Acta 181:723–730
Li YB, Ling LS (2015) Aptamer-based fluorescent solid-phase thrombin assay using a silver-coated glass substrate and signal amplification by glucose oxidase. Microchim Acta 182:1849–1854
Huang HP, Zhu JJ (2009) DNA aptamer-based QDs electrochemiluminescence biosensor for the detection of thrombin. Biosens Bioelectron 25:927–930
Cho HS, Baker BR, Wachsmann-Hogiu S, Pagba CV, Laurence TA, Lane SM, Lee LP, Tok JBH (2008) Aptamer-based SERRS sensor for thrombin detection. Nano Lett 8:4386–4390
Lin ZH, Pan D, Hu TY, Liu ZP (2015) Su XG (2015) a near-infrared fluorescent bioassay for thrombin using aptamer-modified CuInS2 quantum dots. Microchim Acta 182:1933–1939
Cao YL, Shi S, Wang LL, Yao JL, Yao TM (2014) Ultrasensitive fluorescence detection of heparin based on quantum dots and a functional ruthenium polypyridyl complex. Biosens Bioelectron 55:174–179
Wei X, Zhou ZP, Hao TF, Xu YP, Li HJ, Lu K, Dai JD, Zheng XD, Gao L, Wang JX, Yan YS, Zhu YZ (2015) Specific recognition and fluorescent determination of aspirin by using core-shell CdTe quantum dot-imprinted polymers. Microchim Acta 182:1527–1534
Zhang HY, Feng GQ, Guo Y, Zhou DJ (2013) Robust and specific ratiometric biosensing using a copper-free clicked quantum dot–DNA aptamer sensor. Nanoscale 5:10307–10315
Zhang L, Lei JP, Liu L, Li CF, Ju HX (2013) Self-assembled DNA hydrogel as switchable material for aptamer-based fluorescent detection of protein. Anal Chem 85:11077–11082
Sharon E, Freeman R, Willner I (2010) CdSe/ZnS quantum dots-G-quadruplex/hemin hybrids as optical DNA sensors and aptasensors. Anal Chem 82:7073–7077
Wang GL, Hu XL, Wu XM, Li ZJ (2014) Quantum dots-based glucose sensing through fluorescence quenching by bienzyme-catalyzed chromogenic substrate oxidation. Sens Actuators: B 205:61–66
Guo YH, Yao WR, Xie YF, Zhou XD, Hu JM, Pei RJ (2015) Logic gates based on G-quadruplexes: principles and sensor applications. Microchim Acta. doi:10.1007/s00604-015-1633-2
Kong DM, Xu J, Shen HX (2010) Positive effects of ATP on G-quadruplex-hemin DNAzyme-mediated reactions. Anal Chem 82:6148–6153
Zong C, Wu J, Liu MM, Yang LL, Liu L, Yan F, Ju HX (2014) Proximity hybridization-triggered signal switch for homogeneous chemiluminescent bioanalysis. Anal Chem 86:5573–5578
Xue M, Wang X, Wang H, Tang B (2011) The preparation of glutathione-capped CdTe quantum dots and their use in imaging of cells. Talanta 83:1680–1686
Zhang S, Shao YY, Yin GP, Lin YH (2009) Stabilization of platinum nanoparticle electrocatalysts for oxygen reduction using poly (diallyldimethylammonium chloride). J Mater Chem 19:7995–8001
Wu FX, Lewis JW, Kliger DS, Zhang JZ (2003) Unusual excitation intensity dependence of fluorescence of CdTe nanoparticles. J Chem Phys 118:12–16
Yu WW, Qu LH, Guo WZ, Peng XG (2003) Experimental determination of the extinction coefficient of CdTe, CdSe, and CdS nanocrystals. Chem Mater 15:2854–2860
Ge PY, Zhao W, Du Y, Xu JJ, Chen HY (2009) A novel hemin-based organic phase artificial enzyme electrode and its application in different hydrophobicity organic solvents. Biosens Bioelectron 24:2002–2007
Xiao Y, Pavlov V, Gill R, Bourenko T, Willner I (2004) Lighting up biochemiluminescence by the surface self-assembly of DNA-hemin complexes. Chembiochem 5:374–379
Cai K, Lv ZC, Chen K, Huang L, Wang J, Shao F, Wang YJ, Han HY (2013) Aqueous synthesis of porous platinum nanotubes at room temperature and their intrinsic peroxidase-like activity. Chem Commun 49:6024–6026
Dong YQ, Chi YW, Lin XM, Zheng LY, Chen LC, Chen GN (2011) Nano-sized platinum as a mimic of uricase catalyzing the oxidative degradation of uric acid. Phys Chem Chem Phys 13:6319–6324
García-Molina MDM, Muñoz JLM, Martinez-Ortiz F, Martinez JR, García-Ruiz PA, Rodriguez-López JN, García-Cánovas F (2014) Tyrosinase-catalyzed hydroxylation of hydroquinone, a depigmenting agent, to hydroxyhydroquinone: a kinetic study. Bioorg Med Chem 22:3360–3369
Deng C, Chen J, Nie Z, Wang M, Chu X, Chen X, Xiao X, Lei C, Yao S (2009) Impedimetric aptasensor with femtomolar sensitivity based on the enlargement of surface-charged gold nanoparticles. Anal Chem 81:739–745
Xie SB, Chai YQ, Yuan R, Bai LJ, Yuan YL, Wang Y (2012) A dual-amplification aptasensor for highly sensitive detection of thrombin based on the functionalized graphene-Pd nanoparticles composites and the hemin/G-quadruplex. Anal Chim Acta 755:46–53
Xiao LJ, Chai YQ, Yuan R, Wang HJ, Bai LJ (2014) Highly enhanced electrochemiluminescence based on pseudo triple-enzyme cascade catalysis and in situ generation of co-reactant for thrombin detection. Analyst 139:1030–1036
Bang JH, Kamat PV (2009) A tale of two semiconductor nanocrystals: CdSe and CdTe. ACS Nano 3:1467–1476
Zhang LB, Zhu JB, Guo SJ, Li T, Li J, Wang EK (2013) Photoinduced electron transfer of DNA/Ag nanoclusters modulated by G-quadruplex/hemin complex for the construction of versatile biosensors. J Am Chem Soc 135:2403–2406
Lakowicz JR (2006) Principles of fluorescence spectroscopy, 3rd edn. Springer, Baltimore
Wang GL, Jiao HJ, Liu KL, Wu XM, Dong YM, Li ZJ, Zhang C (2014) A novel strategy for the construction of photoelectrochemical sensors based on quantum dots and electron acceptor: the case of dopamine detection. Electrochem Commun 41:47–50
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No.21275065), the Fundamental Research Funds for the Central Universities (JUSRP51314B) and the State Key Laboratory of Analytical Chemistry for Life Science of Nanjing University (KLACLS1008).
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
ESM 1
(DOC 462 kb)
Rights and permissions
About this article

Cite this article
Wang, GL., Hu, XL., Wu, XM. et al. Fluorescent aptamer-based assay for thrombin with large signal amplification using peroxidase mimetics. Microchim Acta 183, 765–771 (2016). https://doi.org/10.1007/s00604-015-1703-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-015-1703-5