
Abstract
The authors describe an electrochemical immunoassay for α-fetoprotein (α-FP) using a glassy carbon electrode (GCE) modified with a nanocomposite made from gold nanoparticles, graphene oxide and multi-walled carbon nanotubes (AuNPs/GO-MWCNTs) and acting as a signal amplification matrix. The nanocomposite was synthesized in a one-pot redox reaction between GO and HAuCl4 without using an additional reductant. The stepwise assembly of the immunoelectrode was characterized by means of cyclic voltammetry and electrochemical impedance spectroscopy. The interaction of antigen and antibody on the surface of the electrode creates a barrier for electrons and causes retarded electron transfer, this resulting in decreased signals in differential pulse voltammetry of hexacyanoferrate which is added as an electrochemical probe. Using this strategy and by working at a potential of 0.2 V (vs. SCE), a wide analytical range (0.01 - 100 ng∙mL‾1) is covered. The correlation coefficient is 0.9929, and the limit of detection is as low as 3 pg∙mL‾1 at a signal-to-noise ratio of 3. This electrochemical immunoassay combines the specificity of an immunological detection scheme with the sensitivity of an electrode modified with AuNPs and GO-MWCNTs.

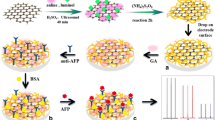
Schematic illustration of the fabrication procedure of the immunosensor
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Accurate and sensitive determination of tumor-related proteins is very important in many modern research fields including biochemistry, biomedicine, and diagnostic [1–3]. α-fetoprotein (α-FP) is an oncofetal glycoprotein with a molecular weight of approximately 70 kDa. It is an important tumor marker and extensively used as clinical cancer biomarkers [4]. The concentration of α-FP is below 25 ng mL−1 in healthy human serum but increases dramatically in the liver cancer patients [5, 6]. Elevated α-FP concentration in serum may be an early indication of some cancerous diseases including yolk sac cancer, hepatocellular cancer, liver metastasis from gastric cancer, nasopharyngeal cancer, and testicular cancer [7, 8]. Thus, sensitive detection of α-FP is absolutely necessary in clinical assay.
There are several methods to detect α-FP, such as electrochemical enzyme immunoassay [9], fluorescence immunoassay [10], enzyme-linked immunosorbent assay [11], chemiluminescence assay [12], and electrochemical assay [13]. Compared with the conventional immunoassay methods, electrochemical immunosensor offers several advantages of high sensitivity and specificity, rapid detection, cost efficiency, low manpower requirements, and inexpensive instrumentation [14].
For an electrochemical immunosensor, its performance is critically dependent on the properties of electrode interface. AuNPs are one of the most widely-used nanomaterials in the fabrication of electrochemical immunosensor for their excellent physicochemical properties including good conductivity and biocompatibility [15, 16]. They can provide more active sites for the binding of antibodies and can accelerate the electron transfer process for signal enhancement. Moreover, the electrocatalytic action of AuNPs facilitates low-potential amperometric measurement of the electrochemical immunosensor [17]. On the other hand, graphene oxide (GO), a representative derivate of graphene, has a large specific surface area with two-dimensional structure. In particular, the presence of abundant carbonyl and carboxyl groups makes GO sheets strongly hydrophilic, allowing them to readily disperse in water and polar organic solvents [18]. There are three advantages when using GO for supporting metal nanoparticles: (i) the rigid two-dimensional structure of GO can enable most surface of the attached nanoparticles to be exposed to the environment, which facilitates nanoparticles show good performance [19]; (ii) metal salts can be reduced by oxygen-functional groups of GO to form metal nanoparticles, avoiding the usage of any toxic reducing reagent [20]; (iii) the strongly hydrophilic of GO can effectively prevent the aggregation of metal nanoparticles. Consequently, GO can be acted as both the reducing agent and the stabilizer for preparation metal nanoparticles. Although the method for metal nanoparticles using GO has so many advantages, the insulating property of GO affects the catalytic activity of the noble metal-GO nanocomposites. Recent studies show that the poor conductivity of GO can be improved after the incorporation of MWCNTs, which serves as an electrical conducting network, increases the basal spacing between the GO sheets and facilitates ionic transportation [21]. Based on the mentioned above, GO-MWCNTs and AuNPs potentially provides an excellent opportunity for signal amplification of electrochemical immunosensors.
We described a simple and clean protocol for anchoring AuNPs on GO-MWCNTs (AuNPs/GO-MWCNTs) by simply mixing GO-MWCNTs with aqueous solutions of HAuCl4. GO acted as both reducing agent and stabilizer, avoiding the use of additional reducing agent and toxic reagents. Further, this nanostructured material has been employed in the development of an electrochemical immunosensor for detection of α-FP, a liver cancer biomarker. The large specific surface area, excellent electron transport capability and strong adsorption capacity of AuNPs/GO-MWCNTs can greatly enhance the electrical signal and improve the immobilizing amount of antibody on the electrode surface. The sensitive detection of α-FP was achieved based on the peak current change of [Fe(CN)6]3-/4- before and after the antigen-antibody reaction. The performance and factors influencing the immunosensor’s performance were investigated in detail. The obtained immunosensor exhibited a good response for the detection of α-FP and showed great potential for application in real sample analysis.
Experimental
Chemicals
α-FP and anti-α-FP antibody were purchased from Bosai Biotechnology co., ltd. (Zhengzhou, China. www.chinabiocell.com). Human serum samples were purchased from a local hospital. Bovine serum albumin (BSA) was obtained from Sigma-Aldrich (www.sigmaaldrich.com). Graphene oxide (GO) was obtained from Nanjing Xianfeng nano Co. (Nanjing, China, www.xfnano.com). HAuCl4 · 3H2O was purchased from Sinopharm Chem. Re. Co. Ltd. (Shanghai, China, www.sinoreagent.com). MWCNTs (purity > 95 %) were purchased from Shenzhen Nanotech Port Co. Ltd. All other reagents were of analytical grade and were used without further purification. Phosphate buffer (0.1 M, pH 7.4) was used as an electrolyte for all electrochemistry measurement. Double distilled water was used throughout the experiments.
Apparatus
The cyclic voltammetric and electrochemical impedance spectroscopy measurements were carried out on a CHI660D electrochemical workstation (Shanghai, China). A standard three-electrode cell contained a platinum wire auxiliary electrode, a saturated calomel reference electrode (SCE) and the modified glassy carbon electrode (GCE) (Ф = 3 mm) as working electrode were employed for electrochemical studies. All potential values given below refer to SCE. Scanning electron microscopy (SEM) analysis was performed using a Hitachi S-3000 N scanning electron microscope.
Preparation of AuNPs/GO-MWCNTs nanocomposites
Two milligrams MWCNTs and 4 mg GO were dispersed in 8 mL double distilled water by ultrasonic agitation for 1 h. Subsequently, 1 mL HAuCl4 aqueous solution (5 mM) was added into the above stable GO-MWCNTs aqueous, and the mixture was stirred for 12 h at 80 °C. Then the resulting nanocomposites of AuNPs/GO-MWCNTs were collected by centrifugation at 2719 g for 20 min and washed with double distilled water for three times. Finally, the precipitation was re-dispersed in 4 mL water and stored in 4 °C for further use.
Fabrication of the immunosensor
The glassy carbon electrode (GCE) was mechanically polished with chamois leather containing 0.05 μm alumina slurry, and then it was ultrasonically cleaned with doubly distilled water, absolute ethanol and doubly distilled water each for 5 min, respectively. 6 μL of the AuNPs/GO-MWCNTs suspension was transferred on the surface of GCE and then dried in air. Then, 5 μL of anti-α-FP solution (200 ng mL−1) was added onto the electrode surface and incubated for 2 h. After washing, 5 μL of 1 wt% bovine serum albumin (BSA) solution was added and incubated for 30 min to eliminate nonspecific binding sites. Subsequently, the electrode was washed and incubated with a varying concentration of α-FP for 40 min at room temperature, and then the electrode was washed extensively to remove unbounded α-FP molecules. The prepared electrode was ready for measurement after washing and the fabricated procedure of the immunosensor was shown in Scheme 1.

Schematic illustration of the fabrication procedure of the immunosensor
Experimental measurements
Electrochemical experiments were carried out in 5 mL phosphate buffer (pH 7.4) containing 5 mM [Fe(CN)6]3-/4- at room temperature. Cyclic voltammetry experiments were recorded at a potential range from - 0.2 to 0.6 V (vs. SCE) with scan rate of 50 mV s−1. Differential pulse voltammetry (DPV) was recorded at a potential from - 0.2 to 0.6 V (vs. SCE) with a pulse period of 0.2 s and amplitude of 50 mV. Electrochemical impedance spectroscopy (EIS) was recorded within a frequency range from 0.1 Hz to 100 kHz, and amplitude of 5 mV.
Results and discussion
Choice of materials
Nowadays, electrochemical biosensors based on metal NPs, including PtNPs, AuNPs, AgNPs, PdNPs and CuNPs, are extensively considered due to their fast and precise response, high sensitivity, simple pretreatment procedures and miniaturizable instrumentation. Among them, AuNPs have drawn much attention in electrochemical field because of its excellent characteristics, such as stable chemical property, favorable biocompatibility, good conductivity, high catalytic activity and good anti-poisoning property [16]. Moreover, owing to their special chemical properties, AuNPs allow a variety of functional groups, including –SH and –NH2, to bind to their surface covalently, which is favorable for the stable immobilization of biomolecules. On the other hand, GO has been of increasing interest for applications in variety fields due to its unique characteristics, such as excellent dispersibility, good biocompatibility and facile surface functionality. Furthermore, GO can successfully reduce the noble metal precursors to metallic NP, forming NP-containing composites, during which GO directly acts as the reductant [20]. However, the insulating property of GO affects the catalytic activity of NP-containing composites. In order to improve its electrical conductivity, we tried to couple the GO with MWCNT through the π–π stacking. And the GO-MWCNTs nanocomposite served as the reducing agent and stabilizer for AuNPs simultaneously. Then the AuNPs/GO-MWCNTs nanocomposite was used to construct an electrochemical immunosensor for α-FP, which can improve the electronic transmission rate as well as increase the surface area to capture a large amount of primary antibodies.
Characterization
The SEM images of GO-MWCNTs and AuNPs/GO-MWCNTs were shown in Fig. 1a and b, respectively. As shown in Fig. 1a, GO with sheet structure was covered by intertwined MWCNTs, resulting in the formation of the percolating network. As to AuNPs/GO-MWCNTs, the SEM analysis (Fig. 1b) indicated a large number of AuNPs with a size of about 200 nm were well-dispersed on the surface of the GO-MWCNTs, which can be attributed to the hydroxyl, epoxide, and carboxylic groups uniformly existing on the GO. A possible mechanism for the formation of the AuNPs/GO-MWCNTs nanocomposites can be attributed to the redox reaction between GO and AuCl4 − [22]. As the structure gave a high specific surface area, it was able to provide a large surface area for antibody attachment.

a SEM image of GO-MWCNTs, b SEM image of AuNPs/GO-MWCNTs, c Raman spectra of the MWCNTs (a), GO (b), AuNPs/GO-MWCNTs (c), d Fourier transform infrared spectroscopy of MWCNTs a, GO (b), AuNPs/GO-MWCNTs (c)
Figure 1c shows the Raman spectra of MWCNTs (a), GO (b) and AuNPs/GO-MWCNTs (c). As shown, all of them displayed two main peaks: the D band and the G band. The D band could be employed to measure the defects of the sample, while the G band could be used to study sp2 carbon networks of the sample. For AuNPs/GO-MWCNTs, the G band located at 1593 cm−1 lower than GO (1606 cm−1), suggested that a larger size of the in-plane sp2 domains were obtained by hybriding GO and MWCNTs, which further confirmed the truth of π–π stacking interaction between GO and MWCNTs [23]. In addition, the intensity of D and G bands for hybrid films increased dramatically in comparison to the GO and MWCNTs, which might be due to the fact the high content of AuNPs in hybrids caused the increase of charge-transfer complexes [24].
Fourier transform infrared spectroscopy of MWCNTs (a), GO (b) and AuNPs/GO-MWCNTs (c) are shown in Fig. 1d. As shown, the pristine MWCNTs did not show any obvious absorption peaks (curve a) over the investigated wavelength range excepted the C = C skeletal vibrations. In the GO spectroscopy (curve b), a broad peak at 3400 cm−1 and a small dip at 3200 cm−1 were observed, which can be ascribed to the intercalated water molecules and O-H stretching vibrations of carboxylic acid group, respectively. The peak at 1625 cm−1 corresponded to the C = C skeletal vibrations of unoxidized graphitic domains. Moreover, the C-O vibrations corresponding to epoxy or alkoxy groups were observed at 1050 cm−1 and the C = O vibrations of the carboxylic acid and carbonyl groups appear at 1736 cm−1 [25]. When the GO formed hybrid with MWCNTs and AuNPs, the characteristic peaks of GO still appeared in the FT-IR spectroscopy (curve c).
Electrochemical characterization of the immunosensor
Cyclic voltammetry (CV) is an effective method for probing the process of electrode modification. Figure 2a showed the CVs of stepwise modified processes of the electrode in the presence of a 5 mM K3[Fe(CN)6]/K4[Fe(CN)6] (1:1) mixture as redox probe in 0.1 M phosphate buffer (pH 7.4) containing 0.1 M KCl. As shown in Fig. 2a, after the pretreated GCE (curve a) was modified with a GO-MWCNTs film (curve b), the peak current density increased. When AuNPs were loaded onto the modified electrode, it can be found that the current density of AuNPs/GO-MWCNTs/GCE (curve c) was further enhanced than that of GO-MWCNTs/GCE (curve b), which was attributed to the excellent electronic transmission ability of AuNPs [26]. When anti-α-FP was immobilized on the electrode surface, a decrease of peak current density was observed (curve d) because the antibody biomacromolecules acting as a nonconductor can obstruct the electron transfer towards electrode surface. After blocked with BSA (curve e) and incubated with α-FP antigen (curve f), the CV responses were declined in succession for the hindrance of BSA and antigen-antibody immunocomplex.

Cyclic voltammograms (a) and electrochemical impedance spectroscopy (b) of the different electrodes: bare GCE (a), GO-MWCNTs/GCE (b), AuNPs/GO-MWCNTs/GCE (c), anti-α-FP/AuNPs/GO-MWCNTs/GCE (d), BSA/anti-α-FP/AuNPs/GO-MWCNTs/GCE (e) and α-FP /BSA/anti-α-FP/AuNPs/GO-MWCNTs/GCE (f)
Electrochemical impedance spectroscopy (EIS) experiments were further performed to probe the features for different layer of electrode modification [27]. The stepwise construction process of the immunosensor was characterized by EIS, and there are significant differences in the impedance spectra for different modified layers of the electrode. As shown in Fig. 2b, the EIS of the bare GCE displayed a small semicircle at high frequencies and linear part at low frequencies (curve a), suggesting very low Ret to redox probe [Fe(CN)6]3-/4-. After the bare electrode was modified with GO-MWCNTs composite film, the resistance for the redox probe decreased due to the electric conducting material of MWCNTs. Moreover, when the composites of AuNPs/GO-MWCNTs were confined on the bare GCE, the resistance for the redox probe further decreased (curve c), implying that AuNPs were excellent electric conducting materials and accelerated the electron transfer. Subsequently, when the anti-α-FP was loaded on the surface of AuNPs, the EIS showed a large increase in diameter (curve d), suggesting that the antibody formed an additional barrier and further prevented the redox probe to the electrode surface. The result was consistent with the fact that the hydrophobic layer of protein insults the conductive support and hinders the interfacial electron transfer. After BSA was used to block non-specific sites, Ret increased in the same way (curve e), which may attribute to the same reason with loading the antibody. Ret further increases (curve f) after the resulting immunosensor was incubated in 100 ng mL−1 of α-FP which indicates the formation of hydrophobic immunocomplex layer embarrassing the electron transfer.
Useful information involving electrochemical mechanism usually can be acquired from the relationship between peak current and scan rate. So, typical CV curves of the resulting immunosensor in 0.1 M phosphate buffer at different scan rates were shown in Fig. 3. It can be seen that a pair of roughly symmetric anodic and cathodic peaks appeared with almost equal peak currents in the scan rate range from 10 to 200 mV s−1. It can be seen that in the inset graph in Fig. 3, the reduction and oxidation peak currents raised linearly with the linear egression equations as i pc = 6.1909 v 1/2 + 10.5354 (R 2 = 0.9960) and i pa = − 5.9496 v 1/2 - 12.0282 (R 2 = 0.9951), respectively, suggesting that the reaction was quasi-reversible diffusion-controlled process.

Cyclic voltammograms of the immunosensor in pH 7.4 phosphate buffer containing 0.1 M KCl and 5.0 mM Fe(CN)6 3-/4- at the different scan rate of (from inner to outer): 10, 20, 40, 60, 80, 100, 120, 140, 160, 180 and 200 mV s−1. The inset shows the linear relationship between the peak currents and the square root of scan rate
The effective surface area and the electron transfer rate of different modified electrodes have been investigated. As can be seen from Fig. S4A (Electronic Supplementary Material), the effective surface area was underpinned by chronocoulometry experiments using 1 mM K3[Fe(CN)6] as model complex, based on Anson equation: Q(t) = 2nFAcD1/2 t1/2/π1/2, where Q is the absolute value of the reduction charge, n is the number of electrons for the reaction, F is the Faraday constant, A is the apparent electrode area, D is diffusion coefficient of the oxidized form, hexacyanoferrate (III), c is concentration of substrate and t is the time. Based on the slopes of the linear relationship between Q and t1/2, the effective surface areas of the GO-MWCNTs/GCE (a) and AuNPs/GO-MWCNTs/GCE (b) were calculated as 0.31 and 0.53 cm2, respectively. The result indicated that the AuNPs/GO-MWCNTs electrode showed larger effective surface area.
The increase in the electron transfer rate was underpinned by experiments of cyclic voltammetry at different modified electrodes of GO-MWCNTs/GCE (a) and AuNPs/GO-MWCNTs/GCE (b) using 5.0 mM [Fe(CN)6]4-/3- as model complex (Fig. S4B). It can be seen that a couple of well defined redox peaks were observed at GO-MWCNTs/GCE (a) with the peak to peak separation (∆Ep) of 123 mV, which corresponded to the quasi-reversible redox behavior of ferricyanide ion. While, on AuNPs/GO-MWCNTs/GCE (b), the ∆Ep value decreased to 109 mV, indicating that sufficiently high conductivity of AuNPs enhanced electron transfer.
Optimization of method
The following parameters were optimized: (a) the concentration of HAuCl4 in the dispersion of GO; (b) sample pH value; (c) temperature; (d) incubation time. Respective data and Figures are given in the Electronic Supplementary Material. The following experimental conditions were found to give best results: (a) the HAuCl4 concentration of 5 mM; (b) a sample pH value of 7.4; (c) a temperature of 25 °C; (d) an incubation time of 40 min.
Analytical performance of immunosensor
To assess the sensitivity and dynamic working range of the electrochemical immunosensor, a differential pulse voltammetry (DPV) measurement was applied to detect α-FP standards in pH 7.4 phosphate buffer containing 5.0 mM Fe(CN)6 3-/4- solution. As can be seen in Fig. 4a, when the α-FP concentration increased, the DPV current signal decreased accordingly. The reason can be attributed to the formation of increasing number of antibody-antigen immunoconjugates, resulting enhanced hindering of electron transfer reaction of the redox probe on the immunosensor surface [28]. It can be seen that the DPV response of the immunosensor decreased with the increment of α-FP concentrations, and exhibited a good linear relationship with the logarithm of α-FP concentration from 0.01 to 100 ng mL−1 (Fig. 4b). The linear regression equation was adjusted to ∆I (μA) = 16.0239 + 6.2879 × log C [α-FP] (ng mL−1, R2 = 0.9929). The relative standard deviations (RSD) for the measurement of each data point were less than 5.0 %. The limit of detection (LOD) was 0.003 ng mL−1 at a signal-to-noise ratio of 3.

Differential pulse voltammetry of the immunosensor after being incubated with different concentrations of α-FP (a–g: 0.00, 0.01, 0.1, 1.0, 10, 50 and 100 ng mL−1) (a). The calibration curve based on the change of the DPV peak currents versus the logarithm of the concentrations (b)
The analytical performance of the immunoassay has been compared with those of other α-FP immunoassays reported (Table 1). The comparative data suggested superiority of the present sensor over some earlier reported methods, especially the detection limit. The wide linear range and low detection limit of the immunosensor might be attributed to two factors: (i) the large specific surface area of AuNPs/GO-MWCNTs, which can increase the sensing surface areas, accordingly more anti-α-FP can be absorbed on the electrode surface, and can enhance the access chance of the antigen and antibody; (ii) the good electron transfer capability of AuNPs/GO-MWCNTs, which greatly enhanced the electrochemical signal.
Selectivity, reproducibility and stability of the immunosensor
In order to assess the selectivity of the immunosensor, interferences study was performed using carcinoembryonic antigen (CEA), human immunoglobulin G (IgG), ascorbic acid and glucose. The peak current after the immunosensor incubated in 10 ng mL−1 of α-FP without or with 100 ng mL−1 of CEA, IgG and 1 mg mL−1 ascorbic acid and glucose were studied (Fig. 5a). Compared with the current response obtained in the presence of only α-FP, the peak current variations from the interferents were less than 5 %. The result indicated that the interference from nonspecific adsorption can be neglected and the selectivity of the immunosensor was thus acceptable.

a Comparison of the response of the immunosensor to 10 ng mL−1 α-FP (1), 10 ng mL−1 α-FP + 100 ng mL−1 CEA (2), 10 ng mL−1 α-FP + 100 ng mL−1 IgG (3), 10 ng mL−1 α-FP + 1 mg mL−1 ascorbic acid (4) and 10 ng mL−1 α-FP + 1 mg mL−1 glucose (4). b The stability of the immunosensor at various storage periods
The reproducibility of the immunosensor was evaluated from the response to 10 ng mL−1 α-FP at five different electrodes. DPV was used to record the electrochemical signal in 5 mM [Fe(CN)6]3-/4- in 10 mM phosphate buffer solution (pH 7.4). The RSD of the measurements for the five electrodes was 4.06 %, suggesting that the reproducibility of the immunosensor was quite good.
The stability of the immunosensor was studied after storage at 4 °C for a 15-day period. The immunosensor was then used to detect 10 ng mL−1 of α-FP. Figure 5b shows the change in current response of immunosensors at various storage times. During the repeated measurement, the activity of immunosensors can maintained at 87.2 % within 15 days, demonstrating a good stability of the immunosensor.
Regeneration of the sensor
The regeneration of the immunosensor was examined by detecting 100 ng mL−1 α-FP with a same immunosensor. After the immunosensor was used to detect α-FP, the electrode was immersed in a bath of base solution containing 0.2 M NaOH and 0.5 M NaCl for 30 min to break the antibody–antigen linkage. The consecutive measurements were repeated six times, and a RSD of 4.8 % obtained. The results demonstrated that the immunosensor could be regenerated and used again.
Real sample analysis
In order to test the analytical reliability and possible application of the designed immunosensor, five human serum samples were assayed using the developed immunosensor. Experimental results were compared with the reference values obtained by the standard ELISA and exhibited in Table 2. The results indicated that there were no significant difference between the two methods, and the RSD was found to be in the range of - 6.93 to 2.32 %, suggesting the immunosensor can be effectively applied to the determination of α-FP in human serum.
Conclusions
In summary, a glassy carbon electrode modified with a nanocomposite of AuNPs/GO-MWCNTs was prepared by a one-pot redox reaction between GO and HAuCl4 without using an additional reductant. Then, a simple electrochemical immunosensor for the detection of α-FP based on AuNPs/GO-MWCNTs composite film was developed. The immunosensor showed excellent selectivity, stability and reproducibility with the detection limit of 0.003 ng mL−1, and showed good accuracy for the α-FP determination in serum samples. The simple fabrication procedure and the high sensitivity demonstrated by the immunosensor may provide many potential applications for the detection of α-FP in clinical diagnostics.
References
Lefkowitz RB, Marciniak JY, Hu CM, Schmid-Schonbein GW, Heller MJ (2010) An electrophoretic method for the detection of chymotrypsin and trypsin activity directly in whole blood. Electrophoresis 31:403–410
Dutsch-Wicherek M (2010) RCAS1, MT, and vimentin as potential markers of tumor microenvironment remodeling. Am J Reprod Immunol 63:181–188
Nie GM, Bai ZM, Yu WY, Chen J (2013) Electrochemiluminescence biosensor based on conducting poly(5-formylindole) for sensitive detection of ramos cells. Biomacromolecules 14:834–840
Wang XW, Xie H (1998) Alpha-fetoprotein enhances the proliferation of human hepatoma cells in vitro. Life Sci 64:17–23
Lin JH, He CY, Zhang LJ, Zhang SS (2009) Sensitive amperometric immunosensor for α-fetoprotein based on carbon nanotube/gold nanoparticle doped chitosan film. Anal Biochem 384:130–135
Jiang W, Yuan R, Chai YQ (2010) Amperometric immunosensor based on multiwalled carbon nanotubes/Prussian blue/nanogold-modified electrode for determination of α-fetoprotein. Anal Biochem 407:65–71
Lin JH, Zhang HH, Niu SY (2014) Simultaneous determination of carcinoembryonic antigen and α-fetoprotein using an ITO immunoelectrode modified with gold nanoparticles and mesoporous silica. Microchim Acta 182:719–726
Nie GM, Li CX, Zhang L, Wang L (2014) Fabrication of a simple and sensitive QDs-based electrochemiluminescence immunosensor using a nanostructured composite material for the detection of tumor markers alpha-fetoprotein. J Mater Chem B 2:8321–8328
Chikkaveeraiah BV, Bhirde A, Malhotra R, Patel V, Gutkind JS, Rusling JF (2009) Single-wall carbon nanotube forest arrays for immunoelectrochemical measurement of four protein biomarkers for prostate cancer. Anal Chem 81:9129–9134
Zheng XT, Li CM (2010) Single living cell detection of telomerase over-expression for cancer detection by an optical fiber nanobiosensor. Biosens Bioelectron 25:1548–1552
Zhou GX, Ireland J, Rayman P, Finke J, Zhou M (2010) Quantification of carbonic anhydrase IX expression in serum and tissue of renal cell carcinoma patients using enzyme-linked immunosorbent assay: prognostic and diagnostic potentials. Urology 75:257–261
Kokado A, Tsuji A, Maeda M (1997) Chemiluminescence assay of alkaline phosphatase using cortisol-21-phosphate as substrate and its application to enzyme immunoassays. Anal Chim Acta 337:335–340
Zhang LY, Yuan R, Huang XQ, Chai YQ, Cao SR (2004) Potentiometric immunosensor based on antiserum of Japanese B encephalitis immobilized in nano-Au/polymerized o-phenylenediamine film. Electrochem Commun 6:1222–1226
Zhang L, Li CX, Zhao D, Wu TT, Nie GM (2014) An electrochemical immunosensor for the tumor marker α-fetoprotein using a glassy carbon electrode modified with a poly (5-formylindole), single-wall carbon nanotubes, and coated with gold nanoparticles and antibody. Microchim Acta 181:1601–1608
Fernández F, Sánchez-Baeza F, Marco MP (2012) Nanogold probe enhanced surface plasmon resonance immunosensor for improved detection of antibiotic residues. Biosens Bioelectron 34:151–158
Li RY, Zhang JJ, Wang ZP, Li ZJ, Liu JK, Gu ZG, Wang GL (2015) Novel graphene-gold nanohybrid with excellent electrocatalytic performance for the electrochemical detection of glucose. Sens Actuators B 208:421–428
Liu X, Li WJ, Li L, Yang Y, Mao LG, Peng Z (2014) A label-free electrochemical immunosensor based on gold nanoparticles for direct detection of atrazine. Sens Actuators B 191:408–414
Yao ZQ, Zhu MS, Jiang FX, Du YK, Wang CY, Yang P (2012) Highly efficient electrocatalytic performance based on Pt nanoflowers modified reduced graphene oxide/carbon cloth electrode. J Mater Chem 22:13707–13713
Xu C, Wang X (2012) Graphene oxide-mediated synthesis of stable metal nanoparticle colloids. Colloid Surf A: Physicochem Eng Asp 404:78–82
Yang MQ, Pan X, Zhang N, Xu YJ (2013) A facile one-step way to anchor noble metal (Au, Ag, Pd) nanoparticles on a reduced graphene oxide mat with catalytic activity for selective reduction of nitroaromatic compounds. CrystEngComm 15:6819–6828
Nie GM, Bai ZM, Chen J, Yu WJ (2012) Simple label-free femtomolar DNA detection based on a nanostructure composite material: MWNT-doped poly(indole-6-carboxylic acid). ACS Macro Lett 1:1304–1307
Wu X, Chai Y, Yuan R, Zhong X, Zhang JJ (2014) Synthesis of multiwall carbon nanotubes-graphene oxide-thionine-Au nanocomposites for electrochemiluminescence detection of cholesterol. Electrochim Acta 129:441–449
Zhang KX, Lu LM, Wen YP, Xu JK, Duan XM, Zhang L, Hu DF, Nie T (2013) Facile synthesis of the necklace-like graphene oxide-multi-walled carbon nanotube nanohybrid and its application in electrochemical sensing of Azithromycin. Anal Chim Acta 787:50–56
Lee JU, Lee W, Yoon SS, Kim J, Byun JH (2014) Site-selective immobilization of gold nanoparticles on graphene sheets and its electrochemical properties. Appl Surf Sci 315:73–80
Zhang C, Ren LL, Wang XY, Liu TX (2010) Graphene oxide-assisted dispersion of pristine multiwalled carbon nanotubes in aqueous media. J Phys Chem C 114:11435–11440
Chen H, Jiang JH, Huang Y, Deng T, Li JS, Shen GL, Yu RQ (2006) An electrochemical impedance immunosensor with signal amplification based on Au-colloid labeled antibody complex. Sens Actuators B 117:211–218
Sun X, Zhu Y, Wang X (2012) Amperometric immunosensor based on deposited gold nanocrystals/4,4’-thiobisbenzenethiol for determination of carbofuran. Food Control 28:184–191
Kavosi B, Hallaj R, Teymourian H, Salimi A (2014) Au nanoparticles/PAMAM dendrimer functionalized wired ethyleneamine–viologen as highly efficient interface for ultra-sensitive α-fetoprotein electrochemical immunosensor. Biosens Bioelectron 59:389–396
Feng DX, Li LH, Han XW, Fang X, Li XZ, Zhang YZ (2014) Simultaneous electrochemical detection of multiple tumor markers using functionalized graphene nanocomposites as non-enzymatic labels. Sens Actuators B 201:360–368
Chen X, Jia XL, Han JM, Ma J, Ma ZF (2013) Electrochemical immunosensor for simultaneous detection of multiplex cancer biomarkers based on graphene nanocomposites. Biosens Bioelectron 50:356–361
Huang KJ, Li J, Wu YY, Liu YM (2013) Amperometric immunobiosensor for α-fetoprotein using Au nanoparticles/chitosan/TiO2–graphene composite based platform. Bioelectrochemistry 90:18–23
Wei Q, Mao K, Wu D, Dai YX, Yang JA, Du B, Yang MH, Li H (2010) A novel label-free electrochemical immunosensor based on graphene and thionine nanocomposite. Sens Actuators B 149:314–318
Shen GY, Zhang XY, Shen YM, Zhang SB, Fang L (2015) One-step immobilization of antibodies for α-1-fetoprotein immunosensor based on dialdehyde cellulose/ionic liquid composite. Anal Biochem 471:38–43
Liang RP, Wang ZX, Zhang L, Qiu JD (2012) A label-free amperometric immunosensor for alpha-fetoprotein determination based on highly ordered porous multi-walled carbon nanotubes/silica nanoparticles array platform. Sens Actuators B 166:569–575
Wang H, Li H, Hang YH, Wei Q, Ma HM (2014) Label-free immunosensor based on Pd nanoplates for amperometric immunoassay of alpha-fetoprotein. Biosens Bioelectron 53:305–309
Acknowledgments
We are grateful to the National Natural Science Foundation of China (grant number: 51302117, 51303073, 51463008), Ganpo Outstanding Talents 555 projects (2013), the Training Plan for the Main Subject of Academic Leaders of Jiangxi Province (2011), the Natural Science Foundation of Jiangxi Province (grant number: 20142BAB206028 and 20142BAB216029), Jiangxi Provincial Department of Education (GJJ11590, GJJ13258), Postdoctoral Science Foundation of China (2014 M551857), Postdoctoral Science Foundation of Jiangxi Province (2014KY14) and the Science and Technology Landing Plan of Universities in Jiangxi province (KJLD12081) for their financial support of this work.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 382 kb)
Rights and permissions
About this article

Cite this article
Gao, YS., Zhu, XF., Yang, TT. et al. Sensitive electrochemical determination of α-fetoprotein using a glassy carbon electrode modified with in-situ grown gold nanoparticles, graphene oxide and MWCNTs acting as signal amplifiers. Microchim Acta 182, 2027–2035 (2015). https://doi.org/10.1007/s00604-015-1537-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-015-1537-1