
Abstract
A method is described for in-situ electrochemical preparation of nanoporous gold electrodes (np-GEs) via alloying and dealloying using ethylene glycol (EG) as a solvent. The np-GEs were prepared via cyclic voltammetry from a gold electrode in the presence of ZnCl2 in EG. Effects of temperature, number of scan cycles and scan rate were investigated. It is concluded that the process involves formation of interfacial elemental zinc and the formation of a gold-zinc alloy. At a temperature of 120 °C, the alloy is formed already after the first scan cycle, and ten further cycles do not cause substantial further changes in the morphology. Changes in the morphology were corroborated by data on the electroactivity of the np-GEs. The mechanism of the formation of np-GEs in EG obviously is different from that in other solvents due to the superior smoothing ability of EG for electroplating. This results in more uniform alloy layers and in a more evenly distributed porous structure. The self-supporting np-GE without any further modification displays an amperometric response to catechol (at a working voltage of 420 mV vs. SCE) in the 0.050 to 1.0 mM concentration range, with a detection limit of 1.78 μM (at an S/N of 3), indicating that the np-GE can be applied to convenient and effective determination of catechol.

A facile in-situ electrochemical alloying and dealloying route using ethylene glycol as the solvent is introduced for the fast fabrication of a self-supporting nanoporous gold electrode. It exhibits good electrocatalytic activity towards the oxidation of catechol.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
As an important metallic nanomaterial, nanoporous gold has been used in catalysis [1–3], electrochemical detection [4–6] and Raman spectroscopy [7, 8]. Its wide applications stimulate the development of an efficient strategy for the fabrication of nanoporous gold. At present, dealloying strategies [9–11] are simpler and more time-saving than template-based methods [12–14]. Compared with chemical alloying/dealloying, which needs tedious procedures and high-temperature apparatus [15], an integrated electrochemical alloying/dealloying is a better strategy [16]. Nanoporous gold can be obtained at a relatively low temperature in a short time without using any hazardous bases or acids [17]. On the basis of the in-situ strategy, a multicyclic electrochemical alloying/dealloying process in benzyl alcohol (BA) [18] or dimethyl sulfoxide (DMSO) [19] at ca. 120 °C was developed to fabricate nanoporous gold electrode (np-GE). The fabrication processes can be completed in one pot under mild conditions without any special protection.
Through the comprehensive analysis, the effects of the preparation conditions on the alloying/dealloying process and the morphology of the gold surface varied widely in different solvents. But the corresponding solvent effects were not clear. In the two solvents, it also needed a long period to obtain an alloy and an np-GE with stable morphology. Therefore, it is necessary to investigate the solvent effect in order to select a more suitable solvent to prepare np-GE facilely. Generally, the solvents suitable for the electrochemical alloying/delloying fabrication of np-GE should meet the following requirements: 1) the solubility of the compound as the precursor of the deposited metal (ZnCl2) should be large enough; 2) the boiling point of the solvent should be higher than the temperature for the direct formation of an alloy on the noble substrate (gold surface); 3) the electrochemical window of the solvent should be wide enough so as not to interfere the preparation of np-GE; 4) the solvent should not be sensitive to water and oxygen so that no special measures are needed for the fabrication process.
Catechol (CC), which common exists in teas, vegetables and traditional medicines, is harmful to humans and animals health [20]. It is imperative to detect catechol conveniently and effectively. A large number of electrochemical biosensors for catechol based on immobilized tyrosinase have been developed [21, 22]. However, these biosensors showed relatively poor long-term stability due to the use of the enzyme. Therefore, nonenzymatic electrochemical sensors with an emphasis put on the development of high-performance electrode materials attract more attention [20]. The traditional nanoporous gold leaf was too thin to be used in electrochemical detection as an independent electrode directly [18] and it was usually adsorbed on a glassy carbon electrode for real application. Using electrochemical alloying/dealloying method, the nanoporous gold layer can be formed directly on the surface of a gold wire, resulting in a so-called self-supporting np-GE and avoiding some bad influence of the poor contact between the nanoporous gold leaf and the substrate electrode on the electron transfer rate. Therefore, the self-supporting np-GE without any modification is more convenient and effective for the determination of catechol.
It has been found that ethylene glycol has strong ability to dissolve large amount of ZnCl2 (as high as 2.0 M ZnCl2 at room temperature), high boiling point (197.3 °C), wide electrochemical window (−2.0 ~ 2.0 V vs. Zn) and oxygen/water-tolerant properties. In addition, similar to the poly(ethylene glycol) [23], ethylene glycol, which is also a smoothing agent for electroplating, has been used in electrodeposition process [24, 25]. This property of ethylene glycol may facilitate formation of a uniform alloy layer and thus well-distributed porous structure in a short time. From this new perspective, ethylene glycol is used to dissolve ZnCl2 as electrolyte in this work, and the solvent effect on the electrochemical alloying/dealloying process and the morphology of np-GE are investigated through the detailed research of the effects of the temperature, the scan cycle and the scan rate. A self-supporting np-GE with high electroactive area is obtained. The electrocatalytic activity of the prepared np-GE towards the oxidation of catechol is also studied and a catechol electrochemical sensor is obtained.
Experimental
Reagents and instruments
Ethylene glycol (A. R.), ZnCl2 (A. R.), 4-aminothiophenol (4-ATP, A. R.) and catechol (A. R.) were purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China, www.reagent.com.cn). Gold wire with 99.999 % purity (0.5 mm in diameter), Zn wire and Zn plate were provided by Tianjin Aidahengsheng Technology Co. Ltd. (Tianjin, China, www.tjaida.cn). ZnCl2/ethylene glycol solution was prepared by dissolving anhydrous ZnCl2 in ethylene glycol at room temperature. All other reagents were of analytical grade and used as received. Triply distilled water (18.25 MΩ cm) was used throughout the experiments.
All the electrochemical experiments were performed on a CHI 630C Electrochemical Analyzer (Chenhua Instrument Co., Ltd., Shanghai, China, www.instrument.com.cn). The surface morphologies of np-GEs were characterized with a JEOL JSM-6700F field emission scanning electron microscopy (SEM) and the element composition were analyzed with an Oxford INCA x-sight energy-dispersive X-ray spectrometer (EDS).
Electrochemical preparation of np-GE
The electrochemical fabrication of np-GEs was carried out in the ZnCl2/ethylene glycol in one pot without deaerating and special caution. The three-electrode system was composed of a working electrode (Au wire), a counter electrode (Zn plate) and a reference electrode (Zn wire). Prior to use, the Au wire was pretreated as follows: firstly, it was immersed in the freshly prepared Piranha solution (3:1 H2SO4 98 % : H2O2 25 %) for 5 min; secondly, the resulting wire was polished with 500 nm and 50 nm alumina slurry on chamois leather; finally, the polished wire was sequentially cleaned in an ultrasonic cleaner with acetone, ethanol and triply distilled water for 5 min, respectively. The geometric area of the gold electrode exposed to the electrolyte was 1.77 mm2. The CV scan range was −0.8 ~ 1.6 V (vs. Zn). When the electrochemical process was over, the gold electrode was taken out and cleaned quickly with ethylene glycol, ethanol and triply-distilled water respectively. Finally, the gold electrode was dried in air.
Electrochemical characterization and measurements
The electrochemical characterization of np-GE was performed in a three-electrode cell composing a working electrode (a smooth gold electrode and np-GEs), a Pt wire counter electrode and a saturated calomel electrode (SCE) reference electrode. The working electrode used in cyclic voltammetric (CV) and amperometric measurements for catechol was a smooth gold electrode or an np-GE prepared after ten CV cycles at 10 mV s−1 at 120 °C. Prior to the measurements, the smooth gold electrode and the np-GEs were cleaned via repeated scan in 0.5 M H2SO4 solution until reproductive curves were obtained. All the measurements were carried out at ca. 30 °C.
Spectroscopic characterization of np-GE
The np-GE prepared after ten CV cycles at 10 mV s−1 at 120 °C was first immersed into a 0.1 mM 4-ATP ethanolic solution for 12 h to construct a self-assembled monolayer. It was then washed thoroughly with ethanol to remove unbound molecules and blown dry with high purity nitrogen. Surface-enhanced Raman scattering (SERS) spectra were recorded on an Ocean Optics QE65000 spectrometer. The excitation wavelength was 785 nm, the laser power was 100 mW, the diameter of the laser spot is 1 μm, and the integration time was 1 s. The spectroscopic characterization was carried out at ca. 30 °C.
Results and discussion
Electrochemical alloying and dealloying of gold electrode
Figure 1 shows the second CV curve of a polished gold electrode in ZnCl2/ethylene glycol at 120 °C. When the potential moved from +1.6 V toward −0.8 V, it was observed that the Zn(II) began to reduce at ca. 0 V and the gold surface became silver gradually, indicating that Zn was deposited on the gold electrode. During the reverse scan, the electrodeposition of Zn continued until 0 V. From 0 V onward, the deposited Zn began to be oxidized (dissolved). It was observed that the electrode surface changed gradually from silver to light red-brown (Fig. S1 in Electronic Supplementary Material, ESM). It can be seen that during the subsequent anodic scan, there are two anodic peaks (a1 and a2) at +0.6 V and +1.0 V, which are assigned to the oxidation of the bulk Zn and the Zn in alloy, respectively [18]. These results indicate that the gold electrode did undergo an alloying/dealloying process. The SEM images of the gold electrode before undergoing the alloying/dealloying process (the inset a in Fig. 1) and the gold electrode obtained after two cycles at 120 °C (the inset b in Fig. 1) show that a porous surface is formed. The formation of the porous structure can be explained with the model described by Erlebacher [26]. When the Zn atoms was selectively dissolved from the outermost alloy layer, the residual Au atoms diffused along the alloy/electrolyte interface and agglomerated into Au-rich islands, exposing the new alloy to the electrolyte. In the process of the continuous dissolution of Zn atoms and the diffusion of the Au atoms, these islands grew to the interconnected porous structure gradually. It was important to note that the Zn was deposited and dissolved at a changing potential when CV method was used. It was seen from the two anodic peaks (Fig. 1) that excess Zn was deposited on the gold surface. So the same amount of alloy was formed in the same time at the same temperature even if the different potential was applied. It was found from the formation mechanism that the dissolution of the inner Zn was more difficult than that of the outer Zn. With the potential shifted positively, the deposited Zn was oxidized gradually from outside to inside and the diffusion time of the Au atoms was prolonged, facilitating the formation of the porous structure.

The second CV curve of a polished gold electrode in ZnCl2/ethylene glycol at 120 °C. Inset: SEM image of the gold electrode before (a) and after (b) two cycles of alloying/dealloying in ZnCl2/ethylene glycol at 120 °C. Scan rate: 10 mV s−1. [ZnCl2] = 1.5 M
Solvent effect on the fabrication and morphology of np-GE reflected by some factors
It is reported that the surface morphology of the resulting np-GE is correlated with the amount of Au-Zn alloy, which depends on the amount of the deposited Zn [17–19]. To understand the solvent effect and its related mechanism during the alloying/dealloying process in ZnCl2/ethylene glycol, it is necessary to investigate the effect of the experiment parameters that directly influence the amount of the deposited Zn and thus the alloy.
Temperature
Figure 2 shows the second CV curves of a polished gold electrode in ZnCl2/ethylene glycol at different temperatures. For the electroreduction of Zn(II), the cathodic current increases with the increase of the temperature due to the lower viscosity and the higher conductivity of the electrolyte at higher temperatures [27]. Therefore, more Zn was deposited on the gold electrode in the same time. For the electrooxidation of Zn, only one anodic peak was observed at temperatures below 70 °C. This peak can be ascribed to the stripping peak of Zn [17–19]. At 70 °C, a new small peak appeared to the right of the main peak (see the amplified curve in Fig. 2b). This shoulder peak is due to the anodic stripping of Zn in the alloy, and its occurrence implies formation of Au-Zn alloy at 70 °C. According to the comparison of the fabrication temperature listed in Table S1 (ESM), this temperature was comparable to that in ZnCl2/DMSO system, but much lower than that in ZnCl2/BA system. Further increase of the temperature made the anodic peak current in the alloy more pronounced, which means that more Au-Zn alloy was formed due to the easier interdiffusion between Au atoms and Zn atoms at higher temperatures.

a The second CV curves of a polished gold electrode in ZnCl2/ethylene glycol at different temperatures. b Amplified curves in a from +0.85 V to +1.25 V. Scan rate: 10 mV s−1. [ZnCl2] = 1.5 M
It’s worth mentioning that the anodic peak potential of Zn at low temperatures was close to that of Zn in the alloy at high temperatures. The unique smoothing ability of ethylene glycol should be responsible for the phenomenon. At lower temperatures, the amount of Zn deposited on the gold electrode was less and the deposited Zn became a thin layer with the help of ethylene glycol. The property of the interfacial Zn (deposited Zn at low temperatures) was very similar to that of the Zn in the alloy, resulting in their similar anodic peak potential. As the temperature increased, the amount of deposited Zn increased, and this true bulk Zn can be oxidized more easily than the interfacial Zn and Zn in the alloy. Therefore, the anodic peak potential of bulk Zn was more negative than the interfacial Zn and Zn in the alloy. The oxidation of Zn in several forms also led to a wider anodic peak at high temperatures. The phenomena described above were not observed in ZnCl2/BA and ZnCl2/DMSO systems. These results indicate that the solvent has an obvious effect on the fabrication process of np-GE.
SEM was used to characterize the surface morphology of the resulting electrodes at different temperatures. Figure 3 shows the SEM images of the electrode after ten CV cycles at different temperatures. It is seen a very smooth surface of the resulting gold electrode at 30 °C (Fig. 3a) and 50 °C (Fig. 3b), indicating Au-Zn alloy wasn’t formed under these conditions. When the temperature increased to 70 °C (Fig. 3c), some shallow cracks resulting from the alloying/dealloying process occur on the gold surface. This result confirms that the aforementioned shoulder peak in the CV curve is the anodic peak of Zn in the alloy. With the further increase of the temperature (from Fig. 3d to Fig. 3f), the pores become larger and the ligaments become thinner, indicating the formation of more obvious bicontinuous structures, which was found at the np-GE obtained in BA but not found at the np-GE obtained in DMSO (Table S1). A color change of the resulting gold electrode, from golden at low temperatures (<50 °C) to dark red-brown at high temperatures, also supports the formation of nanoporous structures. So the temperature is a critical factor affecting the morphology. Higher temperature facilitated the dissolution of Zn atoms and the agglomeration of Au atoms, which were the two important factors for the formation of the bicontinuous structures [27]. The EDS spectrum (Fig. 3g) demonstrated less Zn (3.28 at.%) was remained due to the stronger coordination between Zn(II) and the oxygen-rich ethylene glycol [18, 28], facilitating the dissolution of the Zn atoms and the formation of the bicontinuous porous structure. The corresponding cross-sectional image of the resulting np-GE is shown in Fig. S2 (ESM). Moreover, the uniformity of the np-GE was characterized via the SERS technique. As shown in Fig. S3 (ESM), the present bicontinuous np-GE obtained at 120 °C is uniform on a micrometer scale, indicating it is a good substrate for SERS application.

SEM images of the resulting gold electrode after ten cycles of alloying/dealloying in ZnCl2/ethylene glycol at different temperatures (a-f: 30, 50, 70, 90, 110 and 120 °C). g EDS spectrum of the surface of np-GE obtained at 120 °C. Scan rate: 10 mV s−1. [ZnCl2] = 1.5 M
Scan cycle
Fig. S4 (ESM) shows the CVs of a polished gold electrode in ZnCl2/ethylene glycol at 120 °C under different number of cycles. An anodic peak at +0.95 ~ 1.05 V was observed during the first cycle (Fig. S4a), indicating the electrode had completed a whole alloying/dealloying process. This anodic peak potential was more positive than that of the bulk Zn and the peak had been confirmed to be ascribed to the oxidation of the Zn in the alloy in the previous works [17–19], revealing the formation of the Au-Zn alloy. The corresponding SEM image (Fig. S5a, ESM) shows a coarse surface of gold with a few small and shallow pores due to the short time. After two cycles, the anodic peak current of Zn in the alloy increases and the peak become broader (+0.90 ~ 1.10 V), indicating the formation of more Au-Zn alloy. The corresponding SEM image (Fig. S5b) shows a more obvious porous structure. During the subsequent CV scans, the anodic peak currents of Zn in the alloy are almost the same (Fig. S4b), but the pores become deeper and the ligaments become thinner (Fig. S5c). After ten cycles, the morphology of the resulting gold surface remains almost unchanged (from Fig. S5d to Fig. S5f, ESM), indicating the steady-state of the alloying/dealloying reaction.
The present results are quite different from those in ZnCl2/BA or ZnCl2/DMSO system, where the anodic peak of Zn in the alloy began to appear after five cycles [18] or ten cycles [19] and a well-structured np-GE was obtained after 30 cycles [18] or twenty cycles [19]. The comparison of the fewest scan cycle needed for the initial formation of alloy and the respective final scan cycle for np-GE fabrication was listed in Table S1. It follows that in the present system the time needed for the formation of np-GE is greatly reduced in ethylene glycol. This should be attributed to the smoothing role of ethylene glycol. During the electrodeposition, the formation of the complex (Zn(EG) x 2+) and its adsorption on the gold surface via the lone pair electrons of ethylene glycol [23] reduced the rate of Zn nucleation and avoided the formation of bulky Zn crystal on the gold surface. As a result, a uniform Zn layer (Fig. S6, ESM) and alloy layer were obtained, which in turn facilitated the interdiffusion between Zn atoms and Au atoms.
Scan rate
Fig. S7 (ESM) shows the variation of CV curves of a polished gold electrode with the number of scan cycles at a given scan rate at 120 °C. Compared with a scan rate of 10 mV s−1 (Fig. S4), the charge for Zn reduction decreased obviously at 20 and 30 mV s−1. This indicates that less Zn is deposited on the gold surface in a relatively short time. The increase of the scan rate also shortened the interdiffusion time between Au and deposited Zn, leading to less alloy formed. As seen from the corresponding SEM images of the gold surface after ten cycles, there are no bicontinuous porous structures (Fig. S8a and Fig. S8b, ESM). With the further increase of the scan rate, more cycles are needed for the appearance of the anodic peak current of Zn in the alloy (from the second cycle at 40 mV s−1 (Fig. S7c) to the fifth cycle at 80 mV s−1 (Fig. S7d)). The SEM images show less pores as well as smaller pores (Fig. S8c and Fig. S8d) at higher scan rates.
Fig. S7 also shows that the broad anodic peak of Zn mentioned above splits gradually as the scan rate increases. This interesting phenomenon can be explained as follows. At higher scan rates (>20 mV s−1), less Zn was deposited on the gold surface, and therefore the interfacial Zn predominated and its anodic peak became more obvious accordingly. As a result, the anodic peak of the bulk Zn (ca. +0.6 V) was separated from that of the interfacial Zn (ca. +0.9 V). To further illustrate the effect of the amount of deposited Zn on the change of the anodic peak of the bulk Zn and interfacial Zn, the effect of the ZnCl2 concentration on the alloying/dealloying process was also investigated at 10 mV s−1. Fig. S9 (ESM) shows the second CV curve of a polished gold electrode in ethylene glycol containing different concentrations of ZnCl2 at 120 °C. With the increase of the ZnCl2 concentration, more Zn was deposited on the gold surface, resulting in the relative increase of the percentage of the bulk Zn and the relative decrease of the percentage of the interfacial Zn. As expected, the oxidation of the bulk Zn did become more and more obvious (from curves a to c). This novel phenomenon was not observed in ZnCl2/BA and ZnCl2/DMSO systems. Obviously, this is due to the smoothing role of ethylene glycol for electrodeposition. It follows that different solvents have different effects on the behavior of the electrochemical alloying/dealloying and thus the morphology of the resulting np-GE.
Electroactive area of np-GE
The electroactive area of np-GE is an important aspect of electrode materials for its real application. For quantitative characterization of the electroactive area of np-GEs prepared under different conditions, the CV curves of the np-GEs were recorded in 0.5 M H2SO4 solution. Figure 4 shows a well-defined cathodic peak at ca. +0.9 V (vs. SCE), which results from the electrochemical reduction of gold oxide that was formed during the anodic scan [29]. All the np-GEs have a similar CV curve except the peak current (not shown here). The roughness factor (R f), which is proportional to the electroactive area for a fixed geometric area, is estimated here based on the charge for the reduction of gold oxide [30] (a constant of 386 μC cm−2 is used here to convert the charge to the surface area). The R f values of np-GEs fabricated under different conditions are listed in Table S2 (ESM). It is seen that with the increase of the temperature, the electroactive area of the np-GEs increases markedly, further confirming that higher temperature favors for the alloy formation (the interdiffusion between Au and Zn atoms). With the increase of the scan rate, the electroactive area of the np-GEs decreased markedly, especially from 10 mV s−1 to 20 mV s−1, which corroborates the change of their SEM images (Fig. S8). As for scan cycles, a similar scan cycle dependent morphology change was observed (Fig. S5). It is worth a mention that the electroactive area of the gold electrode is obviously increased after only a few scan cycles, which was not observed in other solvents, further confirming that it is more effective to fabricate np-GE in ethylene glycol. Table S2 also shows that the R f of the np-GEs fabricated after 10, 20 and 30 CV cycles of alloying/dealloying in ZnCl2/ethylene glycol is almost the same, further demonstrating that an np-GE with a high surface area can be obtained in a short time. In addition, the comparison of the R f values of np-GEs fabricated in different solvents was listed in Table S3 (ESM). It can be seen that under the similar conditions, the R f value of np-GE obtained in ethylene glycol was much bigger than that in other two solvents, indicating its better electroanalytical potential. It follows that ethylene glycol is a more suitable solvent for np-GE fabrication via CV technique.

CV curves of the smooth gold electrode (a) and the np-GE (b) in 0.5 M H2SO4 solution at a scan rate of 100 mV s−1. Inset: the magnified curve a. The np-GE was the gold electrode after 10 cycles of alloying/dealloying in ZnCl2/ethylene glycol at 120 °C at a scan rate of 10 mV s−1
Electrocatalytic activity of the np-GE for catechol oxidation
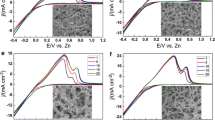
To explore the electrocatalytic activity of the self-supporting np-GE fabricated after ten scan cycles at a scan rate of 10 mV s−1 at 120 °C via CV, the electrochemical measurements were carried out for catechol oxidation. Figure 5a shows the CV grams of a smooth gold electrode and the np-GE in the absence and presence of 0.1 mM catechol in phosphate buffer solution (0.1 M, pH 3.0). It can be seen that the oxidation peak potential of catechol at the np-GE (+0.42 V) is more negative than that at the smooth electrode (ca. +0.7 V). Moreover, the oxidation peak current is much bigger at the np-GE, indicating its good electrocatalytic activity for catechol oxidation. This enhanced electron transfer feature should be due to the high specific surface area and a large amount of catalytic sites of np-GE [2].

a Comparison of the electrocatalytic ability for catechol oxidation at different electrodes. The red curve was obtained at the smooth gold electrode in the blank phosphate buffer solution (0.1 M, pH 3.0). The blue and black curves were obtained at the np-GE in the blank buffer and the buffer containing 0.1 mM catechol, respectively. Scan rate: 50 mV s−1. The used np-GE was fabricated after 10 scan cycles at a scan rate of 10 mV s−1 at 120 °C. b Current response of the smooth gold electrode (red line) at +0.7 V and the np-GE (black line) at +0.42 V on the successive addition of 0.05 mM catechol into the stirred phosphate buffer. Inset: calibration curves of the smooth gold electrode (red line) and the np-GE (black line) in the phosphate buffer containing various catechol concentrations at +0.7 V and +0.42 V, respectively
Figure 5b shows a typical amperometric response of the smooth gold electrode (red line) at +0.7 V and the np-GE (black line) at +0.42 V on the successive addition of 0.05 mM catechol. With the addition of catechol into the stirring solution, the np-GE responded rapidly and a maximum steady-state current was achieved within 10 s. Under the same conditions, however, the response was hardly observed at the smooth electrode. The calibration curve of catechol at the np-GE (see inset in Fig. 5b) shows a good linear line over a catechol concentration range of 0.05 ~ 1.0 mM with the detection limit of 1.78 μM (S/N = 3). The figures of merit of some existing electrochemical methods for catechol determination were listed in Table S4 (ESM). As is shown, the most materials for catechol detection are enzymes, nanoparticles and nanosheets, which are not self-supporting and are usually immobilized on substrate electrodes. The self-supporting np-GE without any modification can detect catechol conveniently and effectively.
Stability, reproducibility and anti-interference
In order to evaluate the operational stability of the present catechol sensor, the amperometric response persistence of the np-GE to catechol (0.1 mM) was investigated in phosphate buffer (pH 3.0) at +0.42 V. As shown in Fig. 6a, the response current is increased rapidly with the addition of catechol and the current is retained about 95.7 % of the initial current at 2000 s. The response current of the sensor to catechol was measured every 3 days and the relative standard deviation (RSD) of the sequential determinations was 3.7 %. Fifty days later, the sensor retained 93.1 % of the initial current. An RSD value of 4.8 % in the amperometric response to 0.1 mM catechol was obtained under the same conditions for five np-GEs prepared in the same way. All the above results indicated that the present catechol sensor had good stability and reproducibility.

a Stability of the amperometric response to 0.1 mM catechol at np-GE in the stirring phosphate buffer (pH 3.0) at +0.42 V. b Amperometric response of np-GE in the stirring phosphate buffer (pH 3.0) at +0.42 V to the subsequent addition of CC (0.1 mM), RS (0.01 mM), HQ (0.01 mM), OPD (0.01 mM), AA (0.01 mM), CC (0.05 mM) and CC (0.1 mM)
The anti-interferential ability of the np-GE was investigated via amperometry for evaluating the potential application of the catechol sensor in real detection. Figure 6b shows the response current of the sensor to catechol and the interferences at +0.42 V in the stirred phosphate buffer (pH 3.0). With each addition of catechol (0.1, 0.05 and 0.1 mM), the sensor responded rapidly. However, the addition of 0.01 mM resorcinol (RS), hydroquinone (HQ), o-phenylenediamine (OPD) and ascorbate acid (AA) didn’t cause obvious current response. These results indicated that the present np-GE had good selectivity to catechol.
Conclusions
A self-supporting np-GE was first prepared in ZnCl2/ethylene glycol via CV. It was found over the tested range, higher temperature, more scan cycles and slower scan rate favored for the formation of bicontinuous porous structure. Due to the smoothing role of ethylene glycol in electroplating, the evolution mechanism of np-GE in ethylene glycol was found to be different from that in other solvents. In ethylene glycol, the interfacial Zn was readily formed on the gold surface, which favors for the formation of Au-Zn alloy. It was found that at 120 °C, Au-Zn alloy can be formed only after one cycle and ten cycles later, the morphology of the resulting np-GE remained almost unchanged. These changes in morphology were confirmed by the quantitative characterization of the electroactive area of the resulting np-GEs prepared under different conditions. It is the smoothing role of ethylene glycol in electroplating that contributes to the formation of uniform alloy layer and thus the well-distributed porous structure. The self-supporting np-GE without any modification has good electrocatalytic activity for catechol oxidation. The present study helps to understand the solvent effects and their related mechanism during np-GE fabrication. It is also helpful for the selection of more suitable solvents to fabricate np-GE using electrochemical alloying/dealloying strategy.
References
Tappan BC, Steiner SA III, Luther EP (2010) Nanoporous metal foams. Angew Chem Int Ed 49:4544–4565. doi:10.1002/anie.200902994
Qiu HJ, Xu CX, Huang XR, Ding Y, Qu YB, Gao PJ (2009) Immobilization of laccase on nanoporous gold: comparative studies on the immobilization strategies and the particle size effects. J Phys Chem C 113:2521–2525. doi:10.1021/jp8090304
Qiu HJ, Zhou GP, Ji GL, Zhang Y, Huang XR, Ding Y (2009) A novel nanoporous gold modified electrode for the selective determination of dopamine in the presence of ascorbic acid. Colloids Surf B 69:105–108. doi:10.1016/j.colsurfb.2008.11.007
Xia Y, Huang W, Zheng JF, Niu ZJ, Li ZL (2011) Nonenzymatic amperometric response of glucose on a nanoporous gold film electrode fabricated by a rapid and simple electrochemical method. Biosens Bioelectron 26:3555–3561. doi:10.1016/j.bios.2011.01.044
Wang GF, He XP, Wang LL, Gu AX, Huang Y, Fang B, Geng BY, Zhang XJ (2013) Non-enzymatic electrochemical sensing of glucose. Microchim Acta 180:161–186. doi:10.1007/s00604-012-0923-1
Chen XM, Wu GH, Cai ZX, Oyama M, Chen X (2014) Advances in enzyme-free electrochemical sensors for hydrogen peroxide, glucose, and uric acid. Microchim Acta 181:689–705. doi:10.1007/s00604-013-1098-0
Lang XY, Guan PF, Zhang L, Fujita T, Chen MW (2009) Characteristic length and temperature dependence of surface enhanced Raman scattering of nanoporous gold. J Phys Chem C 113:10956–10961. doi:10.1021/jp903137n
Li DW, Zhai WL, Li YT, Long YT (2014) Recent progress in surface enhanced Raman spectroscopy for the detection of environmental pollutants. Microchim Acta 181:22–43. doi:10.1007/s00604-013-1115-3
Ding Y, Erlebacher J (2003) Nanoporous metals with controlled multimodal pore size distribution. J Am Chem Soc 12:7772–7773. doi:10.1021/ja035318g
Ding Y, Kim Y, Erlebacher J (2004) Nanoporous gold leaf: “ancient technology”/ advanced material. Adv Mater 16:1897–1900. doi:10.1002/adma.200400792
Ji C, Searson PC (2003) Synthesis and characterization of nanoporous nold nanowires. J Phys Chem B 107:4494–4499. doi:10.1021/jp0222200
Kulinowski KM, Jiang P, Vaswani H, Colvin VL (2000) Porous metals from colloidal templates. Adv Mater 12:833–838. doi:10.1002/(SICI)1521-4095(200006)12:11<833::AID-ADMA833>3.0.CO;2-X
Pu L, Bao XM, Zou JB, Feng D (2001) Individual alumina nanotubes. Angew Chem Int Ed 40:1490–1493. doi:10.1002/1521-3773(20010417)40:8<1490::AID-ANIE1490>3.0.CO;2-K
Meldruma FC, Seshadri R (2000) Porous gold structures through templating by echinoid skeletal plate. Chem Commun 29–30. doi:10.1039/A907074E
Xing XF, Han DQ, Wu YF, Guan YF, Bao N, Xu XH (2012) Fabrication and electrochemical property of hierarchically porous Au-Cu films. Mater Lett 71:108–110. doi:10.1016/j.matlet.2011.12.056
Huang SY, Liu XY, Li QY, Meng MW, Long TF, Wang HQ, Jiang ZL (2010) The preparation of nanoporous gold electrodes by electrochemical alloying/dealloying process at room temperature and its properties. Mater Lett 64:2296–2298. doi:10.1016/j.matlet.2010.07.071
Huang JF, Sun IW (2005) Fabrication and surface functionalization of nanoporous gold by electrochemical alloying/dealloying of Au-Zn in an ionic liquid, and the self-assembly of L-cysteine monolayers. Adv Funct Mater 15:989–994. doi:10.1002/adfm.200400382
Jia FL, Yu CF, Ai ZH, Zhang LZ (2007) Fabrication of nanoporous gold film electrodes with ultrahigh surface area and electrochemical activity. Chem Mater 19:3648–3653. doi:10.1021/cm070425l
Dong H, Cao XD (2009) Nanoporous gold thin film: fabrication, structure evolution, and electrocatalytic activity. J Phys Chem C 113:603–609. doi:10.1021/jp8086607
Huang KJ, Wang L, Li J, Yu M, Liu YM (2013) Electrochemical sensing of catechol using a glassy carbon electrode modified with a composite made from silver nanoparticles, polydopamine, and graphene. Microchim Acta 180:751–757. doi:10.1007/s00604-013-0988-5
Karim MN, Lee JE, Lee HJ (2014) Amperometric detection of catechol using tyrosinase modified electrodes enhanced by the layer-by-layer assembly of gold nanocubes and polyelectrolytes. Biosens Bioelectron 61:147–151. doi:10.1016/j.bios.2014.05.011
Vicentini FC, Janegitz BC, Brett CMA, Fatibello-Filho O (2013) Tyrosinase biosensor based on a glassy carbon electrode modified with multi-walled carbon nanotubes and 1-butyl-3-methylimidazolium chloride within a dihexadecylphosphate film. Sensors Actuators B 188:1101–1108. doi:10.1016/j.snb.2013.07.109
Phuong NV, Kwon SC, Lee JY, Lee JH, Lee KH (2012) The effects of pH and polyethylene glycol on the Cr(III) solution chemistry and electrodeposition of chromium. Surf Coat Technol 206:4349–4355. doi:10.1016/j.surfcoat.2012.04.025
Nguyen HP, Wu M, Su J, Vullers RJM, Vereecken PM, Fransaer J (2012) Electrodeposition of bismuth telluride thermoelectric films from a nonaqueous electrolyte using ethylene glycol. Electrochim Acta 68:9–17. doi:10.1016/j.electacta.2012.01.091
Yamamoto H, Morishita M, Mizuta Y, Masubuchi A (2012) Electrodeposition of Co-Sb thermoelectric film from ethylene glycol-CoCl2-SbCl3 solution. Surf Coat Technol 206:3415–3420. doi:10.1016/j.surfcoat.2012.02.027
Erlebacher J (2004) An atomistic description of dealloying: porosity evolution, the critical potential, and rate-limiting behavior. J Electrochem Soc 151:C614–C626. doi:10.1149/1.1784820
Yang CS, Ma PS, Jing FM, Tang DQ (2003) Excess molar volumes, viscosities, and heat capacities for the mixtures of ethylene glycol + water from 273.15 K to 353.15 K. J Chem Eng Data 48:836–840. doi:10.1021/je020140j
Lee JY, Kim JW, Lee MK, Shin HJ, Kim HT, Park SM (2004) Effects of organic additives on initial stages of zinc electroplating on iron. J Electrochem Soc 151:C25–C31. doi:10.1149/1.1627344
Jiang JH, Wang XY (2012) Fabrication of high-surface nanoporous gold microelectrode. Electrochem Commun 20:157–159. doi:10.1016/j.elecom.2012.04.028
Trasatti S, Petrii OA (1991) Real surface area measurements in electrochemistry. Pure Appl Chem 63:711–734. doi:10.1351/pac199163050711
Acknowledgments
The authors gratefully acknowledge the financial support from the Scientific Research Foundation of Anqing Normal University (140001000029), National Natural Science Foundation of China (21173133) and the National Basic Research Program of China (2011CB707400).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1.25 mb)
Rights and permissions
About this article

Cite this article
Lu, L., Huang, X., Dong, Y. et al. Facile method for fabrication of self-supporting nanoporous gold electrodes via cyclic voltammetry in ethylene glycol, and their application to the electrooxidative determination of catechol. Microchim Acta 182, 1509–1517 (2015). https://doi.org/10.1007/s00604-015-1479-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-015-1479-7