
Abstract
Flower-like gold nanostructures (so called gold nanoflowers) were electrodeposited on indium tine oxide (ITO) glass and served as a SERS-active substrate for sensing dopamine. A double-potential method was applied for the deposition of gold nanostructures on the glass. The density, size, and morphology of the gold nanostructures were controlled by adjusting parameters such as deposition potentials, duration, and concentrations of gold precursors. The particle density on the glass was controlled by changing the overvoltage potentials, while the size and morphology were controlled by changing the concentration of gold precursors and growth times of Gold crystal seeds. The intensity of the Raman spectrum of dopamine was distinctly enhanced on the gold nanostructures with unique flower-like topography, thereby demonstrating the utility of this SERS-active substrate that is facilely obtained by the double-potential deposition method without supporting electrolytes.

Gold nanostructures with optimized flower-like topography are viable SERS substrates to give very distinct Raman spectra of dopamine
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dopamine is a monoamine neurotransmitter associated with proper functioning of the brain, the central nervous system, and the cardiovascular system. Thus, the sensitive and selective detection of abnormal dopamine level in biological fluid is very important in the early detection of several diseases such as schizophrenia, Huntington’s and Parkinson’s disease [1–3]. Common detection methods for neurotransmitters are based on electrochemistry [4–8], chromatography coupled to spectrometry [9–13], fluorescence [14–16] or surface-enhanced Raman scattering [17–21]. The well-known electrochemical oxidation has some limitations due to its low selectivity to dopamine in the presence of many interfering components such as excessive ascorbic acid (AA), uric acid (UP) and epinephrine (EP) [5]. The conventional chromatographic separation also suffers from long preparation times and complicated procedures in spite of high sensitivity [22]. On the other hand, surface-enhanced Raman scattering (SERS) approach has a good potential which can provide both low limits of detection (LOD) and high-level selectivity [17]. The comparable analytical characteristics of various detection methods are summarized in Table 1.
Surface-enhanced Raman scattering (SERS) is a promising spectroscopic analysis which can allow ultrasensitive detection of probe molecules with a clear ‘finger-print’ of the molecule [23]. SERS can magnify the Raman signals of probe molecules adsorbed on roughened metallic surfaces by 106 to 1014 times, allowing the utility of Raman spectroscopy down to the single molecule level [24–27]. Therefore, SERS has received sparkled interests for biomedical applications such as trace analysis, biological diagnosis, and living cell imaging studies [28, 29]. SERS intensities can be tuned by varying the particle size, surface morphology, dielectric environment, and inter–spacing of nanostructures [30]. Among those factors, surface morphology and inter–structure spacing are particularly important because a rough nanostructure surface and close inter–spacing (<5 nm) can enormously enhance the Raman signals of probe molecules through strongly localized electromagnetic fields in those spots.
The main types of the SERS substrates are colloid state and solid-state surface of noble metals. As for colloid state, the random hot-spots are generated by plasmon couplings of closely spaced particles dispersed in solution phase. The resultant SERS activity varies from place to place in colloidal solution, showing a major obstacle in the practical application of SERS [27, 31]. Also, the common method for preparing metal colloids is to reduce metal salts with chemical reducing agents, resulting in the inevitable contamination of the colloids by chemical agents [32]. The presence of chemical species on the metal colloids can give rise to at least two problems: (1) the adsorption of these chemical species on the metallic surface might reduce active adsorption sites for target molecules, and (2) SERS signals originating from these contaminants may severely interfere with the SERS signals of the target molecules, especially when the concentration of the target molecules is very low or the adsorption capability of target molecules is weak.
Hence, the control of homogeneous and density-controlled metallic nanostructures on the solid substrate is in a great demand for obtaining good reproducibility and high sensitivity in SERS measurements. A variety of synthetic methods has been developed to prepare metallic nanostructures with highly textured surface morphology with the use of templates, surfactants or supporting materials [33–36]. The shape–controlled synthesis of metal nanostructures can induce unique optical properties which are strongly dependent on the size and shape of nanostructures [37–40]. Gold nanostructure with a roughened surface morphology on the solid substrate can be considered as a desirable SERS–active substrate with an ultrasensitive detection limit [41–44]. Typically, flower-like nanostructures show high SERS enhancement because of many interstitial sites (or gaps) which can produce extremely intense localized electromagnetic (EM) fields, so called hot spots in SERS measurements [45, 46]. In this regard, electrochemical deposition method can prepare high purity of micro/nanostructures along with well-controlled size, shape, and particle density [47–49]. A double-potential deposition technique is good at controlling metal deposition on the conducting solid surface without using any surfactants and/or supporting electrolytes [50].
SERS technique becomes more popular because a recent advancement of nanotechnology makes it possible to fabricate metal nanostructures with more uniform and roughened morphology. Furthermore, SERS technique has a high-level specificity in a complex media because a specific analyte has its own vibrational “finger-print” spectra [51–53]. In this regard, SERS analysis has many advantages in detecting dopamine molecules as compared to other suitable methods summarized in Table 1. Thus, it is still a hot issue how to prepare the SERS substrates with reproducible and high-quality SERS signals from a specific analyte. Even though colloidal-type SERS substrates can detect dopamine molecules at very low concentration, random hot spots generated by plasmon coupling consequently lead to varying SERS activity that is a major obstacle for applying to SERS-based sensing platform. In contrast, solid-type SERS substrates can directly detect the dopamine molecules with real-time diagnosis.
In this work, we demonstrated the successful synthesis of flower–like Gold nanostructures on the indium tin oxide (ITO) glass as SERS-active substrates using a template-free double–potential method without any templates, surfactants and additional electrolytes. In this method, crystal seeds were formed on the ITO glass by the first pulse with high amplitude, and crystal growth was followed by a small overvoltage of the second pulse without additional nucleation. We systematically investigated the effect of deposition potentials and duration times (i.e., nucleation and growth periods) on the morphology of as–prepared Gold nanostructures and their coupling states. The Raman spectra of dopamine molecules were distinctly enhanced on the optimized Gold nanostructures with flower-like topography, demonstrating a controllable synthesis of SERS–active substrates by the double-potential deposition method.
Experimental
Chemicals
All the chemicals were purchased from Sigma–Aldrich Company: chloroauric acid (HAuCl4), 3–aminopropyl trimethoxysilane (97 %, APTMS), ethanol (99.8 %, C2H5OH), HPLC grade water and dopamine hydrochloride (C6H3(OH)2–CH2–CH2–NH2⋅HCl). All chemicals were of analytical grade and were used without further purification.
Electro-deposition of gold nanostructures
The electro-deposition of Gold nanostructures on the ITO glass with a sheet resistance of 10–15 Ω·cm−2 was performed in a Teflon cell using a standard three-electrode setup. The working electrode was an as-prepared ITO glass, the counter electrode was a Pt wire, and the reference electrode was a saturated calomel electrode (SCE). Gold nanostructures were formed by electrolyzing the aqueous solution of HAuCl4 at different potentials, nucleation times, and growth periods. The operating factors during the electro-deposition process were controlled by a computer–programmed electrochemical workstation at room temperature (CompactStat Plus, HS TECH).
The preparation of Gold nanostructures on the ITO glass is represented by Scheme 1. ITO glass was treated in Plasma Cleaner for 2 min at an RF power of 16.8 W and 150 mTorr of vacuum pressure. Then, it was immediately immersed into APTMS/EtOH (1/9 wt/wt) mixture at 40 °C for 6 h, finally forming APTMS-functionalized ITO glass (i.e., amine-terminated ITO glass). Gold crystals are usually negatively charged [54], and amine–terminated ITO glass is positively charged at pH <9.6 which is the pKa value of terminal amine groups [55]. Thus, negatively charged Gold seeds are easily formed on the positively charged ITO glass at a specified nucleation potential. Then, Gold seeds grow into Gold nanostructures at a given growth potential controlled by the programmed electrochemical workstation. The morphology of Gold nanostructures and their inter–spacing were optimized to offer effective electromagnetic coupling for enhancing SERS signals.

Synthesis of Gold nanostructures on the ITO glass by the double–potential method: 1 Surface activation by an ambient air plasma treatment, 2 ATPMS–functionalized ITO glass substrate, 3 Addition of the first potential to form Gold seeds, 4 Addition of the second potential to grow Gold nanostructures on the ITO glass. Gold-deposited ITO glass is heated at 300 °C for 30 min to remove the residual organics
Characterization
As–prepared samples were characterized by scanning electron microscopy (SEM) and micro Raman spectrometer (ANDOR MOnora500i, 12 mW) to determine the size, morphology, and optical properties of Gold nanostructures deposited on the ITO glass. The Raman spectrograph employed a 1200 g·mm−1 grating and laser excitation at 632.8 nm was used with accumulation time of 5 s.
Results and discussion
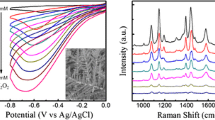
Figure 1 shows the cyclic voltammogram (CV) of pristine ITO and amine–terminated ITO glass scanned from −0.2 V to 1.8 V at 0.01 V·s−1 in 1.0 mM of HAuCl4 solution. A cathodic peak observed at ca. 0.46 V was attributed to the reduction of AuCl4 − to metallic Au0 on the ITO glass by the following reaction: AuCl4 − + 3e− → Au0 + 4 Cl−. According to the CV of pristine ITO glass, a cathodic current flowed up to 0.77 V in the forward scan, which almost coincided with the onset potential of Gold reduction in the reverse scan. According to the CV of amine–terminated ITO glass during a forward scan, a cathodic current flowed up to ca. 0.84 V possibly due to the reduction of Au3+ onto metallic Au0. In other words, the onset of Gold reduction occurred at 0.77 V in the initial reverse scan but continued to flow up to 0.84 V in the forward scan. This hysteresis of onset reduction potentials is called a “nucleation loop” which arises from the over-potential required for nucleation onto the pristine electrode in reference to the Gold deposition onto Gold seeds [49].

Cyclic voltammogram of pristine ITO and APTMS–functionalized ITO glasses in 1.0 mmol·L−1 of HAuCl4 at the scan rate of 0.01 V·s−1
Within the time scale of the reverse scan, the cathodic current due to the reduction of AuCl4 − does not flow until the potential is below 0.77 V even though it is thermodynamically favorable at more positive potentials. This is because an activation barrier must be overcome for the initial reduction of AuCl4 − to form Gold nucleation sites [56]. Once these sites are formed below 0.77 V, the reduction of AuCl4 − can be catalyzed even up to 0.84 V, as shown by the crossover in the CV of amine–terminated ITO glass. Therefore, amine–terminated ITO glass is expected to produce more Gold nanoparticles (NPs) than the pristine ITO glass (data not shown).
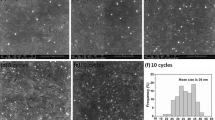
Figure 2 shows the SEM images of Gold nanostructures electrodeposited on amine-terminated ITO glasses prepared at different nucleation potentials (0.4 − 0.8 V) and duration times (2–50 s). Then, HAuCl4 concentration was 1.0 mM and the growth potential (E2) was −0.4 V for 600 s. According to the Fig. 2a, the size and morphology of Gold nanostructures deposited on the ITO glass did not show clear dependence on the nucleation potentials for the constant duration times. The morphology of Gold nanostructures did not also show strong dependence on the duration times at 0.7 V of the nucleation potential as shown in Fig. 2b. Thus, the nucleation potential and duration time were fixed as 0.7 V and 2 s in the following experiments.

a SEM images of Gold nanostructures electrodeposited on amine-terminated ITO glass at the different nucleation potentials (E1) for 2 s: a1 = 0.8 V, a2 = 0.7 V, a3 = 0.5 V. b SEM images of Gold nanostructures electrodeposited on amine-terminated ITO glass at E1 = 0.7 V for different duration times: b1 = 0.5 s, b2 = 2 s, b3 = 50 s, HAuCl4 concentration was 1.0 mM and growth potential (E2) was −0.4 V for 600 s
Figure 3a–c shows the effect of HAuCl4 concentrations on the morphology of Gold nanostructures on the amine–terminated ITO glass. Here, the nucleation potential was 0.7 V for 2 s and the growth potential was −0.4 V for 600 s. HAuCl4 concentrations showed a considerable effect on the formation of Gold nanostructures. When the concentration of HAuCl4 was low (0.5–1.0 mM), flower-like Gold nanoparticles (NPs) of ca. 300–600 nm, so called as Gold nanoflowers (Au NFs), were formed (see Fig. 3a and b). The size of Au NFs increased with HAuCl4 concentration. At a high HAuCl4 concentration of 5 mM, a large dendritic microstructure was formed and sparsely distributed on the ITO glass (see Fig. 3c). At high concentrations of Gold precursors, more Au ions were reduced and formed ad–atoms of Gold on the surface of the ITO glass. Moreover, more ad–atoms of Gold could selectively adsorb on certain crystal faces of small Gold seeds toward specific crystallographic directions, leading to the formation of large Gold crystalline structure [57].

SEM images of Gold nanostructures electrodeposited on amine–terminated ITO glass prepared at different HAuCl4 concentrations: a 0.5 mM, b 1.0 mM, c 5.0 mM, d Raman spectra of dopamines (10−4 M) adsorbed on Gold nanostructures prepared at different HAuCl4 concentrations. The nucleation potential was 0.7 V for 2 s and the growth potential was −0.4 V for 600 s
Figure 3d shows the Raman spectra of dopamines (DOPAs, 10−4 M) obtained from Gold nanostructures electrodeposited on the ITO glass prepared at different concentrations of HAuCl4. The band at ~1480 cm−1 was assigned to the ring stretching vibration, ν19b, attributed to the stretching of the OC-CO bonds. The band at 1270 cm−1 was assigned to the stretching of the catechol C–O bonds. The remaining bands were assigned to various ring stretching vibrations (ν3 = 1325 cm−1, ν19a = 1425 cm−1, ν8a = 1570 cm−1, ν8b = 1590 cm−1) [58]. According to the Raman spectra of DOPAs on Au NFs prepared at 0.5 mM and 1.0 mM of HAuCl4, the latter one exhibited slightly more distinct Raman signals due to its slightly rougher surface morphology. DOPAs on the large Gold crystallites prepared at 5.0 mM of HAuCl4 exhibited very strong Raman signals due to rough surface and large diameter enough to allow the full focusing of a laser beam onto it. Despite strong Raman signals, the non–uniform surface morphology of the dendritic crystallites may not be appropriate for a reliable quantitative analysis in SERS measurements because the Raman spectra of DOPAs were not distinct. In summary, high concentrations of Gold precursors led to large Gold dendrites with non–uniform distribution, whereas low concentration of HAuCl4 produced more densely-packed Gold NFs on the ITO glass with uniform distribution. Thus, appropriate concentration ranges of HAuCl4 are essential to obtain reliable Raman signals of DOPAs from Gold nanostructures deposited on the ITO glass.
Morphological changes of Au NFs prepared at different growth potentials were shown in Fig. 4. At the growth potential of −0.2 V, the amine–terminated ITO glass produced micro–flowers with a size of ca. 1.0 μm (Fig. 4a). At the growth potential of −0.4 V, smaller Au NFs with smooth surface were obtained (Fig. 4b). When the growth potential was further decreased to −0.6 V, spherical-like Gold NPs were obtained with higher particle density (Fig. 4c). The reasons for the variation of particle density are as follows: 1) If the growth potential was not too much below the Nernst equilibrium potential, small Gold seeds dissolved easily due to the negative shift in the reversible potential of the electrode covered by small NPs [59]. Thus, as the growth potential approached the Nernst equilibrium potential, the particle density on the ITO glass decreased. 2) When the growth potential was far below the Nernst equilibrium potential, many Gold nuclei survived without dissolution, increasing particle density on the ITO glass [1]. Morphology changes at different growth potentials can be explained as follows: 1) if the over-potential of the second pulse is too small, Gold can only grow at the lattice plane with the lowest energy of Gold crystallites, leading to the formation of flower-like Au NPs. 2) if the over-potential is large enough, the reaction would have enough energy for Gold to overcome the energy difference between lattice planes. Thus, the Gold nanostructures can grow uniformly in all directions, consequently forming spherical-like Au NPs (Fig. 4c) [43].

SEM images of the Gold nanostructures electrodeposited on amine–terminated ITO glass at different growth potentials: a −0.2 V, b −0.4 V; c −0.6 V for 600 s, d Raman spectra of dopamines (10−4 M) on Gold nanostructures prepared at different growth potentials. HAuCl4 concentration was 1.0 mM and the nucleation potential (E1) of 0.7 V was applied for 2 s
Figure 4d shows the Raman spectra of DOPAs adsorbed on Gold nanostructures prepared at different growth potentials. Gold NFs prepared at the growth potential of −0.2 and −0.4 V exhibited the distinct Raman signals of DOPAs. Even though Au NFs prepared at −0.4 V exhibited lower surface roughness as compared to that of Au NFs prepared at −0.2 V, there was not a significant difference between Raman signals of Au NFs between −0.2 and −0.4 V. The reason may be that Gold NFs prepared at −0.4 V had a higher particle density than that of Au NFs prepared at −0.2 V. Au NFs prepared at −0.6 V exhibited the strong Raman intensity, probably due to the higher particle density. However, the Raman signals of DOPAs were not distinct because of the smoothness of surface morphology. To be conclusive, the optimum SERS substrate for sensing DOPAs can be obtained by adjusting the packing density and surface morphology of Gold NFs deposited on the ITO glass.
Figure 5 shows the morphology variation of Au NFs with increasing growth times. It is obvious that growth times played an important role in determining the final morphology of Gold NFs. At the growth time of 600 s, we observed flower–like crystals (ca. 500 nm), appearing as a clustered form of small Gold NPs (Fig. 5a). When the growth time was prolonged to 1000 s, larger Au NFs with a rougher surface were obtained (Fig. 5b), indicating the continuous growth of the flower–like crystals. Since nucleation was accomplished instantly, the growth times ultimately controlled the packing density and surface morphology of Gold NFs deposited on the ITO glass. Figure 5c shows the comparative Raman spectra of DOPAs (10−4 M) adsorbed on Au NFs prepared by different growth times at the constant growth potential of −0.4 V. Gold nanostructures prepared at the growth time of 1000 s produced more distinct Raman signals of DOPAs than those prepared at the growth time of 600 s, mainly due to the decrease of inter–particle distance and unique flower-like topography, resulting in the effective coupling of Gold nanostructures.

SEM images of the Gold nanostructures electrodeposited on the amine–terminated ITO glass at E2 = −0.4 V for a growth time of 600 s, b growth time of 1000 s. HAuCl4 concentration was 1.0 mM and E1 was 0.7 V for 2 s. c Raman spectra of dopamines (10−4 M) adsorbed on Gold nanostructures prepared at different growth times. Here, E1 and E2 are the nucleation and growth potentials, respectively
In summary, Raman spectra of DOPAs were distinctly enhanced on the optimized Gold nanostructures prepared at the growth potential of −0.4 V for 1000 s. The stretching of the catechol C-O bonds of DOPAs distinctly appeared at 1270 cm−1 (νco). The phenolic stretching vibration was clearly observed at 1480 cm−1 (ν19b), which mainly attributed to the stretching of the C-C bonds to which the oxygen is attached. In particular, ring stretching vibration/deformation mode of OH bond strongly appeared at 1378 cm−1 (ν3/δOH). The remaining bands assigned to various ring stretching vibrations (ν3 = 1325 cm−1, ν19a = 1424 cm−1, ν8a = 1570 cm−1) were observed [60–63]. To be conclusive, the Raman spectra of dopamine molecules were distinctly enhanced on the densely-packed Gold nanostructures with rough flower-like topography.
Conclusions
Flower–like Gold nanostructures, also called Gold nanoflowers (Au NFs), were electrodeposited on the amine–terminated ITO glass using a double–potential electrodepostion method. The main parameters affecting the SERS signals of dopamine were facilely controlled: (1) functionalization of APTMS increased the packing density of Gold nanostructures on the ITO glass, (2) the size, morphology, and packing density of the Gold nanostructures on the ITO glass were controlled by adjusting the HAuCl4 concentrations, deposition potentials, and growth times, and (3) the intensity of Raman spectra of dopamine was distincltly enhanced on the densely-packed Gold nanostructures with unique flower–like topography. The optimized Gold nanostructures produced the distinct Raman spectra of dopmaine molecules, thereby demonstrating the utility of this SERS-active substrate that is easily obtained by template-free double-potential deposition method without any supporting materials.
References
Reisine TD, Fields JZ, Yamamura HI, Bird ED, Spokes E, Schreiner PS, Enna SJ (1977) Neurotransmitter receptor alterations in Parkinson’s disease. Life Sci 21:335–343
Shapiro MG, Westmeyer GG, Romero PA, Szablowski JO, Kuster B, Shah A, Otey CR, Langer R, Arnold FH, Jasanoff A (2010) Directed evolution of a magnetic resonance imaging contrast agent for noninvasive imaging of dopamine. Nat Biotechnol 28:264–270
Lee T, Cai LX, Lelyveld VS, Hai A, Jasanoff A (2014) Molecular-level functional magnetic resonance imaging of dopaminergic signaling. Science 344:533–535
Zheng Y, Huang Z, Zhao C, Weng S, Zheng W, Lin X (2013) A gold electrode with a flower-like gold nanostructure for simultaneous determination of dopamine and ascorbic acid. Microchim Acta 180:537–544
Jackowska K, Krysinski P (2013) New trends in the electrochemical sensing of dopamine. Anal Bioanal Chem 405:3753–3771
Xu T-Q, Zhang Q-L, Zheng J-N, Lv Z-Y, Wei J, Wang A-J, Feng J-J (2014) Simultaneous determination of dopamine and uric acid in the presence of ascorbic acid using Pt nanoparticles supported on reduced graphene oxide. Electrochim Acta 115:109–115
Si P, Chen H, Kannan P, Kim DH (2011) Selective and sensitive determination of dopamine by composites of polypyrrole and graphene modified electrodes. Analyst 136:5134–5138
Wang W, Wang W, Davis JJ, Luo X (2014) Ultrasensitive and selective voltammetric aptasensor for dopamine based on a conducting polymer nanocomposite doped with graphene oxide. Microchimica Acta. doi:10.1007/s00604-014-1418-z
Peterson ZD, Collins DC, Bowerbank CR, Lee ML, Graves SW (2002) Determination of catecholamines and metanephrines in urine by capillary electrophoresis–electrospray ionization–time-of-flight mass spectrometry. J Chromatogr B 776:221–229
Carrera V, Sabater E, Vilanova E, Sogorb MA (2007) A simple and rapid HPLC-MS method for the simultaneous determination of epinephrine, norepinephrine, dopamine and 5-hydroxytryptamine: application to the secretion of bovine chromaffin cell cultures. J Chromatogr B Anal Technol Biomed Life Sci 847:88–94
Ji C, Li W, Ren XD, El-Kattan AF, Kozak R, Fountain S, Lepsy C (2008) Diethylation labeling combined with UPLC/MS/MS for simultaneous determination of a panel of monoamine neurotransmitters in rat prefrontal cortex microdialysates. Anal Chem 80:9195–9203
Zhao XD, Cao YQ, Liu HH, Li FQ, You BM, Zhou XP (2009) Long term high frequency stimulation of STN increases dopamine in the corpus striatum of hemiparkinsonian rhesus monkey. Brain Res 1286:230–238
Syslova K, Rambousek L, Kuzma M, Najmanova V, Bubenikova-Valesova V, Slamberova R, Kacer P (2011) Monitoring of dopamine and its metabolites in brain microdialysates: method combining freeze-drying with liquid chromatography-tandem mass spectrometry. J Chromatogr A 1218:3382–3391
Yildirim A, Bayindir M (2014) Turn-on fluorescent dopamine sensing based on in situ formation of visible light emitting polydopamine nanoparticles. Anal Chem 86:5508–5512
Mu Q, Xu H, Li Y, Ma S, Zhong X (2014) Adenosine capped QDs based fluorescent sensor for detection of dopamine with high selectivity and sensitivity. Analyst 139:93–98
Amjadi M, Manzoori JL, Hallaj T, Sorouraddin MH (2014) Strong enhancement of the chemiluminescence of the cerium(IV)-thiosulfate reaction by carbon dots, and its application to the sensitive determination of dopamine. Microchim Acta 181:671–677
Ranc V, Markova Z, Hajduch M, Prucek R, Kvitek L, Kaslik J, Safarova K, Zboril R (2014) Magnetically assisted surface-enhanced raman scattering selective determination of dopamine in an artificial cerebrospinal fluid and a mouse striatum using Fe(3)O(4)/Ag nanocomposite. Anal Chem 86:2939–2946
Kneipp K, Kneipp H, Itzkan I, Dasari RR, Feld MS (2002) Surface-enhanced Raman scattering and biophysics. J Phys Condens Matter 14:R597–R624
Luo Z, Li W, Lu D, Chen K, He Q, Han H, Zou M (2013) A SERS-based immunoassay for porcine circovirus type 2 using multi-branched gold nanoparticles. Microchim Acta 180:1501–1507
Kaya M, Volkan M (2012) New approach for the surface enhanced resonance Raman scattering (SERRS) detection of dopamine at picomolar (pM) levels in the presence of ascorbic acid. Anal Chem 84:7729–7735
Bu Y, Lee S (2012) Influence of dopamine concentration and surface coverage of Au shell on the optical properties of Au, Ag, and Ag(core)Au(shell) nanoparticles. ACS Appl Mater Interfaces 4:3923–3931
Nandi P, Lunte SM (2009) Recent trends in microdialysis sampling integrated with conventional and microanalytical systems for monitoring biological events: a review. Anal Chim Acta 651:1–14
Kneipp K, Kneipp H, Itzkan I, Dasari RR, Feld MS (1999) Surface-enhanced Raman scattering: a new tool for biomedical spectroscopy. Curr Sci 77:915–924
Kneipp K, Wang Y, Dasari RR, Feld MS (1995) Near-infrared surface-enhanced Raman scattering (NIR-SERS) of neurotransmitters in colloidal silver solutions. Spectrochim Acta A Mol Biomol Spectrosc 51:481–487
Kneipp K, Kneipp H, Itzkan I, Dasari RR, Feld MS (1999) Ultrasensitive chemical analysis by Raman spectroscopy. Chem Rev 99:2957–2976
Kumar GVP, Shruthi S, Vibha B, Reddy BAA, Kundu TK, Narayana C (2007) Hot spots in Ag core − Au shell nanoparticles potent for surface-enhanced Raman scattering studies of biomolecules. J Phys Chem C 111:4388–4392
Luo J, Zeng G-M, Tang L, Yin J, Li Y-P (2009) Study progress on biosensing core/shell nanoparticles. Chin J Anal Chem 37:1847–1852
Cui Y, Ren B, Yao JL, Gu RA, Tian ZQ (2006) Synthesis of AgcoreAushell bimetallic nanoparticles for immunoassay based on surface-enhanced Raman spectroscopy. J Phys Chem B 110:4002–4006
Baron R, Zayats M, Willner I (2005) Dopamine-, L-DOPA-, adrenaline-, and noradrenaline-induced growth of Au nanoparticles: assays for the detection of neurotransmitters and of tyrosinase activity. Anal Chem 77:1566–1571
Yuan W, Li CM (2009) Direct modulation of localized surface plasmon coupling of Au nanoparticles on solid substrates via weak polyelectrolyte-mediated layer-by-layer self assembly. Langmuir 25:7578–7585
Lim DK, Jeon KS, Kim HM, Nam JM, Suh YD (2010) Nanogap-engineerable Raman-active nanodumbbells for single-molecule detection. Nat Mater 9:60–67
Johnson CJ, Dujardin E, Davis SA, Murphy CJ, Mann S (2002) Growth and form of gold nanorods prepared by seed-mediated, surfactant-directed synthesis. J Mater Chem 12:1765–1770
Pan ZW, Dai ZR, Wang ZL (2001) Nanobelts of semiconducting oxides. Science 291:1947–1949
Chen J, Lim B, Lee EP, Xia Y (2009) Shape-controlled synthesis of platinum nanocrystals for catalytic and electrocatalytic applications. Nano Today 4:81–95
Bu Y, Park SJ, Lee S-W (2014) Diamine-linked array of metal (Au, Ag) nanoparticles on glass substrates for reliable surface-enhanced Raman scattering (SERS) measurements. Curr Appl Phys 14:784–789
Bechelany M, Brodard P, Philippe L, Michler J (2009) Extended domains of organized nanorings of silver grains as surface-enhanced Raman scattering sensors for molecular detection. Nanotechnology 20:455302
Wiley B, Sun Y, Chen J, Cang H, Li Z-Y, Li X, Xia Y (2011) Shape-controlled synthesis of silver and gold nanostructures. MRS Bull 30:356–361
Chen J, Wiley BJ, Xia Y (2007) One-dimensional nanostructures of metals: large-scale synthesis and some potential applications. Langmuir 23:4120–4129
Zhang M, Zhao A, Sun H, Guo H, Wang D, Li D, Gan Z, Tao W (2011) Rapid, large-scale, sonochemical synthesis of 3D nanotextured silver microflowers as highly efficient SERS substrates. J Mater Chem 21:18817
Wang H, Halas NJ (2008) Mesoscopic Au “meatball” particles. Adv Mater 20:820–825
Gupta R, Weimer WA (2003) High enhancement factor gold films for surface enhanced Raman spectroscopy. Chem Phys Lett 374:302–306
Yun S, Oh MK, Kim SK, Park S (2009) Linker-molecule-free gold nanorod films: effect of nanorod size on surface enhanced Raman scattering. J Phys Chem C 113:13551–13557
Bu Y, Lee S (2013) Optimizing surface-enhanced RAMAN scattering-active Au nanostructures coated on indium-doped tin oxide glass by combining chemical assembly and electrodeposition methods. Jpn J Appl Phys 52:10MD02
Ye J, Hutchison JA, Uji-i H, Hofkens J, Lagae L, Maes G, Borghs G, Van Dorpe P (2012) Excitation wavelength dependent surface enhanced Raman scattering of 4-aminothiophenol on gold nanorings. Nanoscale 4:1606–1611
Nhung TT, Bu Y, Lee S-W (2013) Facile synthesis of chitosan-mediated gold nanoflowers as surface-enhanced Raman scattering (SERS) substrates. J Cryst Growth 373:132–137
Nhung TT, Lee SW (2014) Green synthesis of asymmetrically textured silver meso-flowers (AgMFs) as highly sensitive SERS substrates. ACS Appl Mater Interfaces 6:21335–21345
Tian Y, Liu H, Zhao G, Tatsuma T (2006) Shape-controlled electrodeposition of gold nanostructures. J Phys Chem B 110:23478–23481
Zhou D-L, Zhang Q-L, Lv Z-Y, Chen W-Y, Liu X-F, Lu Y-H, Wang A-J, Feng J-J (2013) Facile synthesis of a porous network-like silver film for electrocatalytic detection of nitrate. Microchim Acta 180:1495–1500
Ye W, Wang D, Zhang H, Zhou F, Liu W (2010) Electrochemical growth of flowerlike gold nanoparticles on polydopamine modified ITO glass for SERS application. Electrochim Acta 55:2004–2009
Sandmann G, Dietz H, Plieth W (2000) Preparation of silver nanoparticles on ITO surfaces by a double-pulse method. J Electroanal Chem 491:78–86
Halas NJ, Lal S, Chang WS, Link S, Nordlander P (2011) Plasmons in strongly coupled metallic nanostructures. Chem Rev 111:3913–3961
Alvarez-Puebla RA, Liz-Marzan LM (2012) Traps and cages for universal SERS detection. Chem Soc Rev 41:43–51
Wang X, Shi W, She G, Mu L (2012) Surface-Enhanced Raman Scattering (SERS) on transition metal and semiconductor nanostructures. Phys Chem Chem Phys 14:5891–5901
Park S, Park M, Han P, Lee S (2006) The effect of pH-adjusted gold colloids on the formation of gold clusters over APTMS-coated silica cores. Bull Kor Chem Soc 27:1341–1345
Wei S, Wang Q, Zhu J, Sun L, Lin H, Guo Z (2011) Multifunctional composite core-shell nanoparticles. Nanoscale 3:4474–4502
Ivanova OS, Zamborini FP (2010) Electrochemical size discrimination of gold nanoparticles attached to glass/indium-tin-oxide electrodes by oxidation in bromide-containing electrolyte. Anal Chem 82:5844–5850
Qiu C, Shang R, Xie Y, Bu Y, Li C, Ma H (2010) Electrocatalytic activity of bimetallic Pd–Ni thin films towards the oxidation of methanol and ethanol. Mater Chem Phys 120:323–330
Pande S, Jana S, Sinha AK, Sarkar S, Basu M, Pradhan M, Pal A, Chowdhury J, Pal T (2009) Dopamine molecules on aucore − AgshellBimetallic nanocolloids: fourier transform infrared, Raman, and surface-enhanced raman spectroscopy study aided by density functional theory. J Phys Chem C 113:6989–7002
Plieth WJ (1982) Electrochemical properties of small clusters of metal atoms and their role in the surface enhanced Raman scattering. J Phys Chem 86:3166–3170
Bu Y, Lee S (2013) The optical properties of gold nanoparticles with dopamine at different hydrogen ion concentration. J Nanosci Nanotechnol 13:4178–4182
Youn MY, Kim Y, Lee N (1997) Raman spectroscopic study of monodentate dopamine adsorbed on silver and copper adatoms. Bull Korean Chem Soc 18:1314–1316
Lee NS, Hsieh YZ, Paisley RF, Morris MD (1988) Surface-enhanced Raman spectroscopy of the catecholamine neurotransmitters and related compounds. Anal Chem 60:442–446
Volkan M, Stokes D, Vo-Dinh T (2000) Surface-enhanced Raman of dopamine and neurotransmitters using sol–gel substrates and polymer-coated fiber-optic probes. Appl Spectrosc 54:1842–1848
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (20090093908) and by the Korean Government (MEST) (NRF-2010-C1AAA001-2010-0028958).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bu, Y., Lee, S. Flower-like gold nanostructures electrodeposited on indium tin oxide (ITO) glass as a SERS-active substrate for sensing dopamine. Microchim Acta 182, 1313–1321 (2015). https://doi.org/10.1007/s00604-015-1453-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-015-1453-4