
Abstract
We report on a method for the determination of twelve herbicides using solid–liquid–solid dispersive extraction (SLSDE), followed by dispersive liquid-liquid micro-extraction (DLLME) and quantitation by gas chromatography with triple quadrupole mass spectrometric detection. SLSDE was applied to the extraction of herbicides from tobacco samples using multi-walled carbon nanotubes (MWCNTs) as clean-up adsorbents. The effect of the quantity of MWCNTs on SLSDE, and of type and volume of extraction and disperser solvents and of salt effect on DLLME were optimized. Good linearity is obtained in the 5.0 - 500 μg kg−1 concentration range, with regression coefficients of >0.99. Intra-day and inter-day repeatability, expressed as relative standard deviations, are between 3 and 9 %. The recoveries in case of herbicide-spiked tobacco at concentration levels of 20.0, 50.0 and 100.0 g kg−1 ranged from 79 to 105 %, and LODs are between 1.5 and 6.1 μg kg−1. All the tobacco samples were found to contain butralin and pendimethalin at levels ranging from 15.8 to 500.0 μg kg−1.

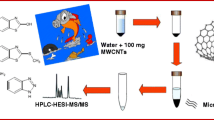
Schematic diagram of herbicide extraction from tobacco samples by SLSDE-DLLME procedures. (a) sample solution containing herbicide and 10 mL acetonitril, (b) MWCNTs cleanup, (c) extract mixed with water, (d) addition of 100 μL of extraction solvent(chloroform) into mixed solution, (e) vortex mixer for 1 min, (f) phase separation after centrifugation. ► A method for analysis of 12 herbicides in tobacco samples was developed. ► MCNTs were used as sorbent, DLLME was further applied to purification and enrichment.. ► Butralin and pendimethalin were found in all tobacco samples.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
During the planting period the tobacco is challenged by various plant diseases, insect pests and weeds, among them various miscellaneous weeds affect the yield of the tobacco. For labor-saving, increasing yield and resistance to disease, herbicides (such as amide herbicide, dinitroaniline herbicide, etc.) are widely used in tobacco production. Although tobacco cannot be eaten directly, many countries still take tobacco as food or quasi food. As absorption of food, herbicide residues could transfer to flue gas with higher delivery rate during the smoking tobacco [1]. Moreover, the intensive application of herbicides has resulted in the contamination of the atmosphere, ground and waste waters, agricultural products and, consequently, in the direct or indirect pollution of food and food products and biological systems, and may represent a serious hazard to human health by environment and the food chain. Hence many countries established maximum recommended limits for herbicide residues in tobacco [2]. China Tobacco Monopoly Bureau at 2002 established its maximum recommended limits for herbicide residues in tobacco, such as, 5.0 mg · kg−1 for butralin and pendimethalin, 20.0 mg · kg−1 for Benfluralin, 1.0 mg · kg−1 for metolachlor, 2.0 mg · kg−1 for diphenamid. Consequently, accurate and reliable methods for the determination of herbicide residues are required for the safety assurance of tobacco products.
Among analytical methods of herbicide residues in tobacco, MS/MS detectors could provide unambiguous identification and accurate determination of herbicide residues at trace levels (<μg L−1), and the detector is particularly suitable to multi-residue analysis of different types of herbicides [3–6]. Nevertheless, most of these techniques require an extensive and time-consuming sample pretreatment.
As is well-known, sample preparation is an important analytical step especially for the determination of trace analytes in complex sample matrices. Although some groups were able to completely omit sample purification by injecting the raw sample extracts [7, 8], unfortunately, the direct injection of raw extracts fails poor sensitivity frequently due to too dirty matrices. Thus, improved sample purification is required. Furthermore, the development of a simultaneous multi-residue extraction method entails difficulties due to the different physicochemical properties of herbicides (polarity, solubility, volatility). Matrix solid-phase dispersion which involves the dispersion of the sample in a solid sorbent, followed by preliminary purification and the elution of the analytes with a relatively low solvent volume and small sample size [9, 10]. The quality of the matrix solid-phase dispersion performance depends on multiple factors, particularly the sorbent type and extraction solvent. Owing to their extremely large surface area and unique structure, multi-walled carbon nanotubes (MWCNTs) can have excellent adsorption ability. In recent reports, CNTs was primarily focusing on the use of SPE method, as a kind of sorbents in a packed column, applied to the extraction of pesticides for water samples [11–15]. Few reports had been published on the use of MWCNTs as a type of solid sorbent materials to absorb the interfering substances in the sample matrices, rather than the analytes [16]. Su et al. [17] used MWCNTs as matrix solid phase dispersion extraction material in butter samples. However, matrix solid-phase dispersion has some disadvantages, such as too many manual operating steps. Thus, the development of a simple, rapid and low organic solvent-consuming sample preparation procedure combining extraction and cleanup in one single step is of great interest. For this purpose, a novel one-step sample preparation technique, namely ultrasound or microwave-assisted solid–liquid–solid dispersive extraction (SLSDE) [18, 19]. During the extraction procedure, the analytes are extracted into the extraction solvent while the interfering matrix components are retained by the dispersing sorbent.
In addition, it was necessary for the preconcentration to improve the detection limit of target analytes. Recently, a novel microextraction technique, dispersive liquid–liquid microextraction (DLLME), has been reported for extracting and/or preconcentration target analytes from samples [20–24]. DLLME has high preconcentration capabilities in a very short time. The main disadvantage of the DLLME is that it cannot apply to directly extraction of solid substances and heavily contaminated extracts. Thus, in order to overcome this problem it is necessary to include a clean-up stage before DLLME technique. Dispersive solid-phase extraction was recently introduced as a rapid and simple technique for clean-up crude extracts of different food and environmental samples [25, 26]. Compared to dispersive solid-phase extraction technology, SLSDE technology may be more rapid and simple.
In this work, we describe a novel and accurate method for the determination of herbicide residues in tobacco with SLSDE procedure and DLLME procedure. Efficiency of SLSDE extractions were optimized by comparing with the amount of sample, type and quantity of sorbents, and nature and volume of the extraction solvent. The parameters affecting the DLLME procedure such as type and volume of extraction and disperser solvents, salt effect and extraction time were studied. Moreover, the method performance was evaluated by studies of recovery, limits of detection, analysis of real samples.
Experimental
Reagents and materials
Analytical standards of benflurain, clomazone, acetochlor, alachlor,metolachlor, butralin, pendimethalin, diphenamid, butachlor, benfluralin, napropamide, nitrofen were supplied by J&K Technology Co. Ltd. (Beijing, China, www.jkchemical.com). Sodium chloride (NaCl), bromobenzene (C6H5Br), carbon tetrachloride (CCl4), chlorobenzene(C6H5Cl), chloroform, 1,2-dichloroethane (C2H4Cl2), dichlorometane (CH2Cl2), methanol, acetonitrile, and acetone, all of analytical grade, were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China, reagent360.cn.b2b168.com). Ultrapure water was obtained from a Milli-Q system from Millipore (Milford, MA, USA) and used throughout the work.
MWCNTs with average external diameters of 10–20 nm, 5 nm i.d. and PSA were provided by Tianjing Agela Co. Ltd. Co. (Tianjing, China, www.agela.com.cn). All sorbent types were first washed three times with n-hexane and then three times with methanol. MWCNTs were dried for 2 h at 120 °C to remove the absorbed water and then kept it in desiccators for storage.
Samples
Tobacco samples were provided by China tobacco Fuzhou Industrial Corporation. The samples were dried naturally, then, samples were finely milled using a knife and homogenized to achieve a representative samples and then dispensed into plastic bags. All samples were stored at 4 °C and out of direct sunlight until the analysis. Spiked samples were prepared by adding standard solution to 1 g of blank samples. The spiked samples were mixed on a vortex mixer for 1 min and then incubated at room temperature for 2 h before analysis.
Extraction procedure
A diagram of the SLSDE-DLLME protocol was shown in Fig. 1. The homogenized tobacco sample (1 g) was extracted with 10 mL of a mixture of acetonitrile. MWCNTs (200 mg) were added into the mixture. Then, the mixture was mixed on a vortex mixer for 20 s, and sonicated for 10 min. After centrifuged for 3 min at 4,500 rpm, the upper phase was used for the DLLME process.

Schematic diagram of herbicide extraction from tobacco samples by SLSDE-DLLME procedures. a sample solution containing herbicide and 10 mL acetonitril, b MWCNTs cleanup, c extract mixed with water, d addition of 100 μL of extraction solvent(chloroform) into mixed solution, e vortex mixer for 1 min, f phase separation after centrifugation
Chloroform (100 μL) as DLLME extraction solvent was added to an aliquot of 1 mL of acetonitrile extract, which used as disperser solvent, and the mixture was rapidly injected into a 15 mL screw cap glass centrifuge tube with conical bottom containing 4 mL of water. The ternary component system was mixed by vortex mixer for 1 min. The cloudy solution (water, acetonitrile, and chloroform) stably formed for a long time in the test tube. After centrifugation for 3 min at 4,500 rpm, most of the supernatant was removed with a Pasteur pipette and the sedimented chloroform phase was quantitatively transferred to a small vial using a 200 μL intubation.
Instruments and chromatographic conditions
An Agilent 7890A GC system (Agilent Technologies, Palo Alto, CA, USA) equipped with an autosampler (Agilent 7683) was coupled to a 7000A QqQ mass detection, operating in electron impact ionization mode. The GC separation was performed using a DB-1701 capillary column with a length of 30 m × 0.25 mm I.D. and a film thickness of 0.25 μm (J&W Scientific, USA). Temperature program: 40 °C holds for 1 min, 30 °C min−1 up to 130 °C, 5 °C min−1 up to 250 °C, 20 °C min−1 up to 270 °C, hold for 2 min. The total running time was 31 min. The temperatures for the injection port, transfer line and ion source were set at 280 °C, 280 °C and 230 °C, respectively, and a solvent delay of 10 min was selected. Splitless injection of 1 μL sample was carried out. Helium was used as carrier gas at a flow rate of 1.2 mL min−1. Electron impact mass spectra were measured in the range from m/z 50 to m/z 400 at acceleration energy of 70 eV. The system was operated in MS/MS mode using nitrogen as collision gas at a flow rate of 1.5 mL min−1 and using helium as quenching gas at a flow rate of 2.25 mL min−1 in the collision cell. The retention times, precursor ions, product ions and collision energy for identification and quantitation of 12 herbicides were shown in Table 1.
Calibration curve and evaluation of method performance
For method validation, five point matrix matched calibration curves were obtained by spiking blank tobacco samples with the target herbicides in the range from 5.0 to 500.0 μg kg−1, and each spiking level was prepared two times.
Limits of detection (LOD) and of quantification (LOQ) were determined using blank tobacco samples spiked at herbicides levels with 20.0 μg kg−1. Each level was processed in triplicate by optimized method, and the LODs and LOQs were calculated by extrapolation of the concentrations giving a signal-to-noise ratio of 3 and 10, respectively.
For recovery studies, the tobacco samples were spiked at three concentration levels 20.0, 50.0, and 100.0 μg kg−1 for herbicides. All the experiments were carried out in triplicate.
Results and discussion
SLSDE extraction process
Efficiency of SLSDE extractions depends on the amount of sample, type and quantity of sorbents, nature and volume of the extraction solvent. To obtain the high recoveries and low interferences, the parameters that affected the partition of analytes were optimized such as the amount and type of solid phase sorbent.
The analytes were extracted by 10.0 mL acetonitrile, and further cleaned up and dried by mixing with the MWCNTs sorbents and anhydrous MgSO4. The MWCNTs cleanup step was designed to retain matrix components and allow the analytes of interest into the acetonitrile phase. To evaluate the effect of this parameter, different amounts of MWCNTs were investigated in the same SLSDE extraction process. The amount of sorbent was increased from 50.0 to 200.0 mg. As shown in Table 2, by increasing the amount of MWCNTs from 50.0 mg to 200.0 mg, the recoveries for most herbicides remained at the acceptable level (79.2–90.8 %). However, the recoveries decreased to 6–12 % when the amount of MWCNTs was increased from 200.0 mg to 400.0 mg, partial herbicides were adsorbed on the MWCNTs. In addition, although better recoveries were achieved with 50 mg materials, the cleanup performance was not as good as 200.0 mg. The recoveries could be acceptable at the amount of 200 mg MWCNTs. Consequently, 200.0 mg (10.0 mL extract) was used as the optimum amount for the SLSDE extraction process in the further studies since acceptable recoveries and good cleanup performances were obtained at this amount.
PSA is typically used as SLSDE sorbents to remove the interfering substances. However, sometimes the PSA-cleanup performance is not good enough to remove the interfering substances in the matrices. The tobacco sample processed by MWCNTs looked transparent in color and the PSA-cleanup sample had deeper color. MWCNTs displayed a better cleanup performance than PSA to remove pigment in tobacco. Compared to the use of PSA, the application of MWCNTs extended the lifetime of liner, which also showed the less contamination with MWCNTs cleanup.
The solvent used to extract the analytes from matrix must then act as disperser in DLLME process, therefore, its selection must take into account both the properties required to the extraction. Methanol, acetonitrile, and acetone are normally used as disperser solvents in DLLME. In order to select the extraction solvent, spiked samples were extracted with each selected extraction solvent and the obtained extracts were processed by DLLME. The same DLLME process was performed on the blank tobacco extracts, spiked with herbicides after extraction step, to evaluate separately the efficiency of the extraction and DLLME steps. The recoveries of the whole procedure (SLSDE + DLLME) and DLLME procedure of all extraction solvents tested. All extraction solvents were able to efficiently extract herbicides from samples with the recoveries ranging from 70 % to 110 %. However, methanol and acetone produced co-extracted matrix components.
Optimization of DLLME
To obtain the optimal DLLME conditions for the determination of herbicides in tobacco, the influence of different experimental parameters on DLLME performance (type and volume of extraction solvent, salt addition and water volume) were carefully investigated.
The type of extraction solvent is one of the most important parameters affecting the DLLME efficiency. In a preliminary optimization step, several halogenated solvents, with density higher than water and different polarities, were proved as possible DLLME extraction solvent. Most of them (CH2Cl2, C6H5Br, C6H5Cl and C2H4Cl2) were found insoluble in dispersive solvent at volumes above 20–30 μL, so only CHCl3 and CCl4 were evaluated. For both solvents, a stable cloudy solution was observed. CCl4 showed lower extraction efficiency than to chloroform (Fig. 2), thus the latter was selected as DLLME extraction solvent.

Effect of extraction solvent on the extraction efficiencies of herbicides. Volume of extraction solvent: 100 μL; percentage of NaCl, 0 %; spiked herbicide concentration: 50.0 μg kg-1. Herbicide No. 1, Benflurain; 2, Clomazone; 3, Acetochlor; 4, Alachlor; 5, Metolachlor; 6, Butralin; 7, Pendimethalin; 8, Diphenamid; 9, Butachlor; 10, Benfluralin; 11, Napropamide; 12, Nitrofen
Volume of extraction solvent and percentage of NaCl were tested, and their high and low levels were chosen according to preliminary experiments. The volume of extraction solvent to be added in order to obtain the highest extraction efficiency of the analyte was studied within a volume range of 50–200 μL. 50 μL of extraction volume was completely dissolved in the aqueous bulk. It is possible to observe that the greater relative extraction efficiency for herbicides was obtained when 100 μL chloroform based on peak signal response and the extraction phase volume. By increasing the volume of chloroform from 100 μL to 200 μL, the extraction phase volume increased and the recoveries of herbicides decreased due to a dilution effect of it. Therefore, 100 μL chloroform was selected in order to obtain higher recovery and lower detection limit. The effect of ionic strength on the efficiency of micro-extraction was evaluated by adding NaCl in the range of 0–10 %. The DLLME experimental conditions were the same as those described before. The results showed that by increasing the amount of NaCl, recoveries were not varied. Moreover, no salt was added in the further experiments due to low salting out effect in this study. As a consequence, DLLME was carried out without salting.
To study the effect of the dispersive solvent volume on extraction efficiency, different volumes of SLSDE extract, from 0.5 mL to 3.0 mL with gaps of 1.0 mL, were added to different volumes of deionized water from 2.0 mL to 6.0 mL, respectively. With the increase of deionized water ratio, the extraction efficiency firstly increased and then decreased, and the volumes of sedimented phase obtained were gradually increased. Thus, 1.0 mL of the SLSDE extract and 4.0 mL of deionized water were chosen as the optimal volume for the dispersive solvent and deionized water.
Regarding the optimal DLLME conditions, the experimental factor values extrapolated by the experimental design study, to obtain the highest responses, were as follows: 100 μL of chloroform, 1.0 mL of the SLSDE extract, 4.0 mL of H2O and no addition of NaCl. According to these results, the optimal conditions selected for the DLLME step were employed for the rest of experiments in this study.
Analytical performance
The linear range of the method was established using blank tobacco samples spiked with the target compounds at five levels from 5.0 to 500.0 μg kg-1 for the herbicides, with five calibration levels, each injected in triplicate with good correlation coefficients of more than 0.99. The typical chromatograms of the tobacco DLLME extracts before and after spiking with herbicides were reported in Fig. 3. It can be seen, some interfering peaks were observed in the elution region of the analytes for tobacco matrices, however, these interference peaks were not found near the retention time of the analytes in the EI MRM CID Figure.

GC-MS/MS chromatogram of 12 herbicides in a blank tobacco sample spiked a concentration of 50 μg kg−1 (a) and a typical blank tobacco sample (b). 1, Benflurain; 2, Clomazone; 3, Acetochlor; 4, Alachlor; 5, Metolachlor; 6, Butralin; 7, Pendimethalin; 8, Diphenamid; 9, Butachlor; 10, Benfluralin; 11, Napropamide; 12, Nitrofen
The LODs ranged from 1.5 to 6.1 μg kg−1 while the LOQs ranged from 5.0 to 15.0 μg kg−1 (Table 2). For recovery studies, blank tobacco samples were spiked with herbicides at three concentration levels of 20.0, 50.0 and 100.0 μg kg−1, and The intraday recoveries obtained ranged from 79 % to 105 % at all spiked levels, while the intreday recoveries obtained ranged from 76 % to 100 % at all spiked levels. The intraday repeatability of the method expressed as relative standard deviations (RSDs) for six replicates ranged from 3 % to 7 %, while the inter-day repeatability of the method expressed as RSDs for six replicates ranged from 5 % to 9 % (Table 3).
Application of the method to real samples
To evaluate the methodology, 100 tobacco samples from the Fuzhou city were collected and detected. All the samples were extracted and analyzed by the optimal method. Butralin and pendimethalin were detected in all the samples, and their concentrations were ranged between 15.8 and 499.1 μg kg−1. As for high and serious residual amount, the possible reason was that butralin and pendimethalin as systemic suckercides directly smeared tobacco leaf buds at the late growth of tobacco. Diphenamid ranging between 7.6 and 21.3 μg kg−1 were analyzed in the 85.8 % of the samples, while clomazone ranging between 5.2 and 9.8 μg kg−1 were analyzed in the 56.3 % of the samples. Diphenamid and clomazone belonged to prenatal systemic herbicide, thus they could be detected with less residual amount. Acetochlor was rarely used in tobacco production, but 3.8 % of the samples ranging between 6.5 and 405.0 μg kg−1 were still detected, for wheel crops such as rice, vegetables may use it. No other herbicides were found, probably due to less use or less than the detection limit.
Conclusions
In this work, a fast and simple preparation method was developed and evaluated for the analysis of twelve herbicides in the tobacco samples. MWCNTs were proved to be a new and effective sorbent and were successful applied for detection of herbicides at trace levels in sample cleanup. The SLSDE method realized the simultaneous extraction and purification, and as for alternative method of matrix solid-phase dispersion, which reduced manual factors. The enrichment step has been considerably simplified by introducing the DLLME procedure, in which the extractive solvent mixed with the extract is simply dispersed in deionized water being the analytes concentrated by centrifugation in the sedimented phase. The DLLME procedure as a concentration step showed time and expenses saving are and high enrichment effect. The proposed method met the requirements for herbicide analysis (average recovery values were in the range 79–105 % for all selected herbicides with RSD values lower than 9 %). So many herbicide residues were detected, showed the feasibility of the method.
References
IARC monographs (186) On the evaluation of the carcinogenic risk of chemicals to humans. Tobacco Smoking IARC.Lyon,France.1886,38
Davis DL, Nielsen MT (1999) Tobacco: production, chemistry and technology [M]. Blackwell Science Limited, Oxford
Lee J, Park J, Jang G, Hwang K (2008) Comparative study of pesticide multi-residue extraction in tobacco for gas chromatography-triple quadrupole mass spectrometry. J Chromatogr A 1187:25–33
Haib J, Hofer I, Renaud JM (2003) Analysis of multiple pesticide residues in tobacco using pressurized liquid extraction, automated solid-phase extraction clean-up and gas chromatography-tandem mass spectrometry. J Chromatogr A 1020:173–187
Mayer-Helm B (2009) Method development for the determination of 52 pesticides in tobacco by liquid chromatography-tandem mass spectrometry. J Chromatogr A 1216:8953–8959
Mayer-Helm B, Hofbauer L, Müller J (2008) Method development for the determination of selected pesticides on tobacco by high-performance liquid chromatography-electrospray ionisation-tandem mass spectrometry. Talanta 74:1184–1190
Yang S, Ding J, Zhu L, Hu B, Li J, Chen H, Zhou Z, Qiao X (2009) Detection of melamine in milk products by surface desorption atmospheric pressure chemical ionization mass spectrometry. Anal Chem 81:2426–2436
Zhang JL, Li Z, Zhang CS, Feng BS, Zhou ZG, Bai Y, Liu HW (2012) Graphite-coated paper as substrate for high sensitivity analysis in ambient surface-assisted laser desorption/ionization mass spectrometry. Anal Chem 84:3296–3301
Beyer A, Biziuk M (2008) Applications of sample preparation techniques in the analysis of pesticides and PCBs in food. Food Chem 108:669–680
Gilbert-López B, García-Reyes JF, Molina-Díaz A (2009) Sample treatment and determination of pesticide residues in fatty vegetable matrices: a review. Talanta 79:109–128
Wang S, Zhao P, Min G, Fang G (2007) Multi-residue determination of pesticides in water using multi-walled carbon nanotubes solid-phase extraction and gas chromatography–mass spectrometry. J Chromatogr A 1165:166–171
Zhou Q, Wang W, Xiao J (2006) Preconcentration and determination of nicosulfuron, thifensulfuron-methyl and metsulfuron-methyl in water samples using carbon nanotubes packed cartridge in combination with high performance liquid chromatography. Anal Chim Acta 559:200–206
Zhou Q, Xiao J, Wang W, Liu G, Shi Q, Wang J (2006) Determination of atrazine and simazine in environmental water samples using multiwalled carbon nanotubes as the adsorbents for preconcentration prior to high performance liquid chromatography with diode array detector. Talanta 68:1309–1315
El-Sheikh AH, Sweileh JA, Al-Degs YS, Insisi AA, Al-Rabady N (2008) Critical evaluation and comparison of enrichment efficiency of multi-walled carbon nanotubes, C18 silica and activated carbon towards some pesticides from environmental waters. Talanta 74:1675–1680
El-Sheikh AH, Insisi AA, Sweileh JA (2007) Effect of oxidation and dimensions of multi-walled carbon nanotubes on solid phase extraction and enrichment of some pesticides from environmental waters prior to their simultaneous determination by high performance liquid chromatography. J Chromatogr A 1164:25–32
Zhao P, Wang L, Zhou L, Zhang F, Kang S, Pan C (2012) Multi-walled carbon nanotubes as alternative reversed-dispersive solid phase extraction materials in pesticide multi-residue analysis with QuEChERS method. J Chromatogr A 1225:17–25
Su R, Wang X, Xu X, Wang Z, Li D, Zhao X, Li X, Zhang H, Yu A (2011) Application of multiwall carbon nanotubes-based matrix solid phase dispersion extraction for determination of hormones in butter by gas chromatography mass spectrometry. J Chromatogr A 1218:5047–5054
Zhong Z, Li G, Wu Y, Luo Z, Zhu B (2012) Ultrasound-assisted matrix solid-phase dispersive liquid extraction for the determination of intermediates in hair dyes with ion chromatography. Anal Chim Acta 752:53–61
Zhou T, Xiao X, Li G (2012) Hybrid field-assisted solid–liquid–solid dispersive extraction for the determination of organochlorine pesticides in tobacco with gas chromatography. Anal Chem 84:420–427
Rezaee M, Assadi Y, Milani Hosseini MR, Aghaee E, Ahmadi F, Berijani S (2006) Determination of organic compounds in water using dispersive liquid-liquid microextraction. J Chromatogr A 1116:1–9
Adlnasab L, Ebrahimzadeh H, Yamini Y (2012) A three phase dispersive liquid-liquid microextraction technique for the extraction of antibiotics in milk. Microchim Acta 179:179–184
Alothman ZA, Yilmaz E, Habila M, Shabaka A, Soylak (2013) Ligandless temperature-controlled ionic liquid-phase microextraction of lead(II) ion prior to its determination by FAAS. Microchim Acta 180:669–674
Rahimi-Nasrabadi M, Zahedi MM, Pourmortazavi SM, Heydari R, Rai H, Jazayeri J, Javidan A (2012) Simultaneous determination of carbazole-based explosives in environmental waters by dispersive liquid—liquid microextraction coupled to HPLC with UV–vis detection. Microchim Acta 177:145–152
Rabieh S, Bagheri M, Planer-Friedrich B (2013) Speciation of arsenite and arsenate by electrothermal AAS following ionic liquid dispersive liquid-liquid microextraction. Microchim Acta 180:415–421
Mol HG, Plaza-Bolaños P, Zomer P, de Rijk TC, Stolker AA, Mulder PP (2008) Toward a generic extraction method for simultaneous determination of pesticides, mycotoxins, plant toxins, and veterinary drugs in feed and food matrixes. Anal Chem 80:9450–9459
Drozdzynski D, Kowalska J (2009) Rapid analysis of organic farming insecticides in soil and produce using ultra-performance liquid chromatography/tandem mass spectrometry. Anal Bioanal Chem 394:2241–2247
Acknowledgments
This work was supported by the fund from Fuzhou Tobacco Company of Jiangxi province and key laboratory for quality and safety of agricultural products.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liao, Q.G., Zhou, Y.M., Luo, L.G. et al. Determination of twelve herbicides in tobacco by a combination of solid–liquid–solid dispersive extraction using multi-walled carbon nanotubes, dispersive liquid-liquid micro-extraction, and detection by GC with triple quadrupole mass spectrometry. Microchim Acta 181, 163–169 (2014). https://doi.org/10.1007/s00604-013-1086-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-013-1086-4