
Abstract
We have developed a highly sensitive electrochemical immunoassay for the quantitation of zearalenone (ZEN), a mycotoxin produced by Fusarium species. In this enzyme linked immunosorbent assay, the enzymatic conversion of the substrate p-nitrophenylphosphate is detected by a microplate reader and the signal subsequently converted into an electrical signal. The concentrations of coating antigen (ZEN-ovalbumin), of monoclonal antibody, and of goat anti-mouse antibody labeled with alkaline phosphatase were optimized. In terms of electrochemical detection, the types and pH values of the buffers, the conditions for agitating, and scanning frequency were optimized. The effective detection range of this immunoassay is quite wide (0.004 to 9.5 ng mL−1), and the limit of detection is 2 pg mL−1. ZEN-free corn, wheat, and grain-based food samples were spiked with ZEN and analyzed by this method, and recoveries were found to range from 91.6 % to 113.0 %. Unlike previously described electrochemical methods, this method is both highly sensitive and has a wide working range. The method is fast and thus provides a platform for high-throughput analysis that meets the current need to monitor trace levels of analytes in grain and grain-based food.

Scheme of test procedure of electrochemical immunosensor (procedure of immune-reaction: from a to f)
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Zearalenone (ZEN), a secondary metabolites from Fusarium species, is a mycotoxin found throughout the world, and primarily in grain crops, such as corn, wheat, barley, and oats [1, 2]. Classified as a nonsteroidal estrogen or mycoestrogen [3] with immune toxicity [4] and reproductive toxicity [5] towards humans and animals, ZEN has been causally linked to abortion and stillbirth in livestock. To decrease the incidence of ZEN-based toxicity towards humans, European Commission regulations specify a maximum level of 20 μg of ZEN per kg of cereal-based foods intended for consumption by infants and young children [6]. This upper limit is considerably lower than the maximum amount reported in cereals (100 μg.kg−1) for adults. The need for more stringent control of ZEN levels in the food of infants and young children has necessitated urgent measures to develop highly sensitive, rapid, and simple instrumentation, with a wide dynamic range in order to monitor the content of the ZEN in food samples.
Increasing expectations for the highly sensitive detection for mycotoxins in food samples has led to the development of many traditional analytical methods, such as gas chromatography–mass spectrometry [7], high-performance liquid chromatography-fluorescence detection [8], high-performance liquid chromatography-mass spectrometry [9], enzyme linked immunosorbent assay (ELISA) [10], radioimmunoassay [11], and fluorescent immunoassay [12]. Nonetheless, these methods still did not meet the current trace level and high-throughput demands simultaneously for the detection of ZEN in certain foodstuffs.
Electrochemical sensors respond to specific analytes by converting the signal generated by a chemical reaction to an electrical signal. Owing to the high sensitivity of electrochemical sensing methods, different kinds of electrochemical analysis have been reported, including methods that involve either glass carbon electrodes [13] or screen-printed electrodes [14]. However, these methods require immobilization of antigen conjugates or antibodies on the electrodes that provide a solid phase for the immune reaction, and this immobilization reduces the generation of electrochemical signals and hinders electron transfer [15]. Another type of electrochemical immunoassay, which uses magnetic beads, enables highly sensitive detection of ZEN in baby food [15, 16], although the narrow detection range cannot be adjusted to detect different levels of sample contamination.
Alkaline phosphatase (ALP) converts the colorless substrate p-nitrophenylphosphate (pNPP) to the yellow product p-nitrophenol (pNP), which can be detected by a microplate reader set to monitor the change in 405 nm of optical density (OD). Therefore, the level of an analyte in a sample is proportional to the OD405 nm value, and can be quantified using a calibration curve prepared using an analyte standard. Given that pNP has electrochemical activity, whereas pNPP does not, residual unreacted pNPP in the substrate buffer does not affect the detection of pNP. This permitted the development of electrochemical analytical method based on the catalytic conversion of pNPP for the detection of drug residues [17]. In initial studies, sensitivity was insufficient to detect trace amounts of analyte, and the working range was narrow because each electrochemical analysis could not be coupled with other methods, such as ELISA. Moreover, this approach has not yet been used to detect mycotoxins, for which there is an urgent need for a rapid, highly sensitive method with a wide dynamic range.
In this study, a highly sensitive electrochemical immunoassay that involved pNP was established. Combined use of ELISA and electrochemical analysis improved the sensitivity and working range of the approach to enable large-scale detection. Advantages of the strategy relative to the current alternatives are that it is highly sensitive and has a wider detection range, enabling high throughput, rapidity, and accuracy. This new platform permits trace detection across a wide working range for analysis of food samples.
Materials and methods
Reagents
Whereas ZEN and pNPP were purchased from the Sigma Chemical Co. (St. Louis, MO, USA, http://www.sigmaaldrich.com), ovalbumin (OVA) and dry skimmed milk were obtained from Sangon Biotech (Shanghai, China, http://www.sangon.com/), Tween 20 was obtained from Generay Biotech (Shanghai, China, http://www.generay.com.cn/), a specific monoclonal antibody against ZEN (2C9) was prepared in our laboratory [18], and ALP-labeled goat anti-mouse antibody was obtained from ProteinTech (Chicago, IL, USA, http://www.ptglab.com/).
Equipment
Microtiter plates were purchased from Nunc (Roskilde, Denmark, http://www.nuncbrand.com), the microplate reader was obtained from Bio-Tek (Winooski, VT, USA, http://www.biotek.com/) and the CHI630C multifunction-voltammograph and three-electrode systems were obtained from Chenhua (Shanghai, China, http://www.chinstr.com/).
Development and optimization of electrochemical immunoassay
After conjugation of ZEN and OVA as described previously [19], the ZEN-OVA conjugates were diluted in 0.05 M carbonate buffer (pH 9.6) and used as the coating antigen. The 96-well microplate was coated with the conjugates for 4 h at 37 °C, and blocked using skimmed milk for 2 h at 37 °C. We added 100 μL of a mixture of diluted monoclonal antibody and standard solution of ZEN (1:1, v/v) to the wells after they were washed three times with 0.01 M phosphate buffer saline (PBS, pH 7.4) containing 0.05 % Tween-20 (PBST). Plates were then incubated for 1 h at 37 °C. After washing the plates with PBST, ALP-labeled goat anti-mouse antibody was added to the wells, and the plates were incubated for 1 h at 37 °C. After washing with PBST, the substrate (pNPP) solution was added, and the plates were incubated for 30 min at 37 °C before adding stop solution. The plates were then transferred to the microplate reader to record absorbance values, and then analyzed using the three-electrode system provided with the multifunction-voltammograph work station.
Different concentrations of coating antigen (ZEN-OVA), monoclonal antibody, and ALP-labeled goat anti-mouse antibody were assessed by checkerboard titration involving ELISA and electrochemical methods to improve sensitivity [16]. In order to enhance the performance of electrochemical analysis, the reaction buffers, such as Britton-Robison buffer (BR), Tris–HCl, and borate buffer (BB) with different pH values (1.0, 3.0, 5.0, 7.0, and 9.0) were investigated. Different conditions of agitating (0.1 V, 0 V, -0.1 V for 30 s, 90 s, and 180 s respectively) and scanning frequency were also determined to improve the sensitivity of electrochemical analysis.
Sample preparation
The ZEN-free corn and wheat samples and grain-based food samples (noodles and biscuit) were milled and provided by the Shanghai Entry-Exit Inspection and Quarantine Bureau. To spike and extract the samples, standard ZEN solution (in methanol) was added to 5 g samples at different levels (0.2, 1, 2, 10, 20, 100, and 200 μg.kg−1), then homogenized and allowed to stand at room temperature overnight. First, each 5 g sample was added to 25 ml methanol:H2O (80:20, v/v), and shaken vigorously for 15 min at room temperature. Second, the supernatants were purified by centrifugation at 2000 g for 10 min. Finally, extracts were diluted five times in 0.01 M PBS before subsequent analysis.
Test procedure
First, 96-well microplates were coated with ZEN-OVA conjugates (diluted 1/4,000) for 4 h at 37 °C, and blocked using 5 % skimmed milk for 2 h at 37 °C. Extracts of spiked samples or standard solutions of ZEN (50 μL, range from 0 to 50 ng.mL−1) were then mixed with the same volume of monoclonal antibody (50 μL, 1/81,000), and incubated for 1 h at 37 °C. The mixtures (100 μL) were then added in the wells of microtiter plates, which had already been washed with 0.01 M PBST three times. The plates were incubated for 1 h at 37 °C, and washed with 0.01 M PBST three times. Then, 100 μL of ALP-labeled goat anti-mouse antibody (1/2,000) was added to the plates and incubated for 1 h at 37 °C. After washing three times, the substrate solution (100 μL of 10 mM pNPP in 0.1 M diethanolamine buffer, containing 1 mM MgCl, pH 9.8) was added, and the plates were incubated for 30 min at 37 °C. 50 μL of 2.0 M NaOH was added to terminate reaction. Finally, the plates were analyzed by transfer to a microplate reader to record the OD405 nm of the contents of the wells. Samples (100 μL) from each well were added to 6 mL BR buffer (0.2 M, pH 3.0), and agitated for 90 s in -0.1 V. The parameters used for the electrochemical immunoassay included the scanning range (from 0.9 to 1.5 V), pulse amplitude (50 mV), pulse duration (20 s), pulse period (200 s) and scanning speed (50 mV.s−1), as described previously [17]. The oxidation peak current was observed by differential pulse voltammetry (DPV) using the CHI630c work station. Each sample was evaluated three times in duplicate. The scheme of the test procedure is shown in Fig. 1.

Scheme of test procedure of electrochemical immunoassay (procedure of immune-reaction: from a to f)
Results and discussion
Optimization of the electrochemical immunoassay
Concentrations of ZEN-OVA, ZEN-specific monoclonal antibody, and ALP-labeled goat anti-mouse antibody needed to be optimized using ELISA and electrochemical analysis, because each detection method has a different detection range, and could not detect all concentrations of ZEN in the range from 0 to 50 ng.mL−1. The optimum concentrations of coating antigen (1/4,000, 0.25 μg.mL−1), monoclonal antibody (1/81,000, 0.12 μg.mL−1) and ALP labeled goat anti-mouse antibody (1/2,000, 0.5 μg.mL−1) were determined by checkerboard titration performed through ELISA and electrochemical analysis. For the electrochemical part of this immunoassay, several parameters were optimized to improve the performance of detection. First, the effects of using different reaction buffers (BR, Tris–HCl, and BB) were determined. The BR buffer was chosen on the basis that it could conduct more current than either Tris–HCl or BB and did not have the base peak. The Tris–HCl buffer had an oxidation peak current at 1.20 V, whereas the pNP peak was at 1.2–1.4 V. Thus, Tris–HCl could affect the current of the pNP when used as the reaction buffer. For BB, the current associated with pNP was not stable at a given concentration.
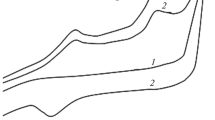
Next, the effects of modifying the pH of BR buffer were investigated. The oxidation peak currents of pNP when different pH values of BR buffer (pH values of 1.0, 3.0, 5.0, 7.0, and 9.0) were tested, are shown in Fig. 2. The current was largest at pH 3.0. Different conditions of agitating were also determined. The results indicated that 90 s at –0.1 V is the largest current (data not shown). The optimum scanning frequency was determined by seven continuous scans. The oxidation peak currents recorded are shown in Fig. 3. Data from the first scan was used, because it had a larger current than recorded using the other scans.

Oxidation peak currents of pNP at different pH values of BR buffer

Oxidation peak currents of pNP of scanning frequencies curves (from a to g, the scanning frequencies were 1, 2, 3, 4, 5, 6, and 7 times)
Calibration curves for the detection of ZEN
The calibration curves for the ELISA and electrochemical parts of the immunoassay are shown in Figs. 4 and 5. For ELISA (y = –0.4418× + 0.6776,R2 = 0.9921), as the concentrations that gives 20 % and 80 % inhibition (IC 20-IC 80) of color development, the detection range was from 0.35 to 9.5 ng.mL−1, EC50 (concentration for 50 % of maximal effect) was 2.7 ng.mL−1. Based on the concentration, which results in a 10 % inhibition (IC 10) of color development, the limit of detection (LOD) was 0.16 ng.mL−1. For the electrochemical analysis (y = –0.1558× + 0.46, R2 = 0.9943), the detection range was from 0.004 to 0.55 ng.mL−1, EC50 was 0.054 ng.mL−1, and the detection limit was 0.002 ng.mL−1.

Calibration curve of ELISA for the immunoassay

Calibration curve of electrochemical analysis for the immunoassay
According to the overlap in the detection ranges of the ELISA and electrochemical components, the detection range of our immunoassay ranged from 0.004 to 9.5 ng.mL−1, with a detection limit of 0.002 ng.mL−1. Considering the dilution ratio (5 × 5) during sample extraction, the limit of detection was 0.05 μg.kg−1, and the working range was 0.1–237.5 μg.kg−1 in samples. The relative standard deviations (RSD) were below 10 % for both ELISA and electrochemical analysis.
Recovery studies
Levels of ZEN in the spiked corn and wheat samples, and grain-based food samples (0.2, 1, 2, 10, 20, 100, and 200 μg.kg−1) were determined using the electrochemical immunoassay. The results are shown in Table 1. Each sample was evaluated three times in duplicate to verify the repeatability. The recovery rates range from 91.6 % to 113.0 %, indicating the accuracy of this method in both grain and grain-based food samples.
Based on the overlap concentrations (0.4 and 0.5 ng.mL−1) of dynamic ranges in the two different detection methods and the dilution ratio of sample extraction (5 × 5), ZEN-free samples (corn, wheat, noodles and biscuit) were spiked in 10 and 12.5 μg.kg−1. The spiked samples were determined by ELISA and electrochemical analysis, respectively. The results were shown on Table 2. For both methods the recovery rates were almost identical and statistical analyses using Pearson correlation by SPSS software (11.5 version) show a significant correlation at the P < 0.01 level between the two methods. The recovery rates for detection of ZEN in samples have a good correlation between the two different methods, which means the dynamic ranges obtained from two different detection methods overlap.
Comparison of different methodologies for determination of ZEN
An outline of recent studies about ZEN detection is presented in Table 3. These methods were mainly immunoassays with the advantages of rapidity and accuracy. High sensitivity (0.011 and 0.007 ng.mL−1) was reported, albeit with narrow working ranges [15, 16]. A method that used surface plasmon resonance involved a complicated procedure with additional clean-up steps was not suitable for detection by various farms or companies, although it had an even wider dynamic working range [20].
Owing to their rapidity and sensitivity, sensors based on colorimetry [21], fluorescence [12], and electrochemistry [15, 16, 22] have been employed in recently published detection methods. Along with the wider applications of sensors in analytical methods and increased concerns about food safety, the need for higher sensitivity and a wider dynamic range has become a particularly important request. In this study, the highly sensitive detection capacity (0.002 ng.mL−1) and wide working range (0.004–9.5 ng.mL−1) of our immunoassay was achieved by combining ELISA and electrochemical analysis, which only involved an additional 1–2 min of assay time compared with electrochemical analysis alone [16].
Conclusions
In this study, a highly sensitive electrochemical immunoassay with a wide dynamic working range was developed for the detection of ZEN in foodstuffs. The limit of detection was 0.002 ng.mL−1, which could ensure detection of trace amounts of ZEN in grain and grain-based foods. The detection range (0.004–9.5 ng.mL−1) was wider than most broadly comparable detection methods, and the approach is compatible with high-throughput analysis, saving time, labor, and money.
References
Placinta CM, D’ Mello JPF, Macdonald AMC (1999) A review of worldwide contamination of cereal grains and animal feed with Fusarium mycotoxins. Anim Feed Tech 78:21–37
Tanaka K, Sago Y, Zheng Y, Nakagawa H, Kushiro M (2007) Mycotoxins in rice. Int J Food Microbiol 119:59–66
Bennett JW, Klich M (2003) Mycotoxins. Clin Microbiol Rev 16:497–516
Vlata Z, Porichis F, Tzanakakis G, Tsatsakis A, Krambovitis E (2006) A study of zearalenone cytotoxicity on human peripheral blood mononuclear cells. Toxicol Lett 165:274–281
Collins TF, Sprando RL, Black TN, Olejnik N, Eppley RM, Alam HZ, Rorie J, Ruggles DI (2006) Effects of zearalenone on in utero development in rats. Food Chem Toxicol 44:1455–1465
European Commission Regulation (EC) no. 856/2005 of 6th June 2005, amending Regulation (EC) no. 466/2001 as regards Fusarium toxins (2005). Off J Eur Union 143:3–8
Kinani S, Bouchonnet S, Bourcier S, Porcher JM, Ait-Aissa S (2008) Study of the chemical derivatization of zearalenone and its metabolites for gas chromatography–mass spectrometry analysis of environmental samples. J Chromatogr A 1190:307–315
Aresta A, Palmisano F, Vatinno R, Zambonin CG (2006) Ochratoxin a determination in beer by solid-phase microextraction coupled to liquid chromatography with fluorescence detection: a fast and sensitive method for assessment of noncompliance to legal limits. J Agric Food Chem 54:1594–1598
Soleimany F, Jinap S, Abas F (2012) Determination of mycotoxins in cereals by liquid chromatography tandem mass spectrometry. Food Chem 130:1055–1060
Burmistrova NA, Goryacheva IY, Basova EY, Franki AS, Elewaut D, Van Beneden K, Deforce D, Van Peteghem C, De Saeger S (2009) Application of a new anti-zearalenone monoclonal antibody in different immunoassay formats. Anal Bioanal Chem 395:1301–1307
Thouvenot D, Morfin RF (1983) Radioimmunoassay for zearalenone and zearalanol in human serum: production, properties, and use of porcine antibodies. Appl Environ Microb 45:16–23
Chun HS, Choi EH, Chang HJ, Choi SW, Eremin SA (2009) A fluorescence polarization immunoassay for the detection of zearalenone in corn. Anal Chim Acta 639:83–89
Owino J, Arotiba O, Hendricks N, Songa E, Jahed N, Waryo T, Ngece R, Baker P, Iwuoha E (2008) Electrochemical Immunosensor Based on Polythionine/Gold Nanoparticles for the Determination of Aflatoxin B1. Sensors 8:8262–8274
Bonel L, Vidal JC, Duato P, Castillo JR (2010) Ochratoxin A nanostructured electrochemical immunosensors based on polyclonal antibodies and gold nanoparticles coupled to the antigen. Anal Methods-UK 2:335–341
Hervás M, López MÁ, Escarpa A (2010) Simplified calibration and analysis on screen-printed disposable platforms for electrochemical magnetic bead-based inmunosensing of zearalenone in baby food samples. Biosens Bioelectron 25:1755–1760
Hervás M, López MÁ, Escarpa A (2009) Electrochemical immunoassay using magnetic beads for the determination of zearalenone in baby food: An anticipated analytical tool for food safety. Anal Chim Acta 653:167–172
Song WW, Ding MX, Zhang NW, Liu HF, Xu MG, Liu GY, Chai CY (2007) Immuno-voltammetric Determination of Chloramphenicol Residues in Milk. Chinese J Anal Chem 35:1731–1735
Wang YK, Wang J, Wang YC, Chen ZF, Yan YX, Hao QW, Li SQ, Yu C, Yang CY, Sun JH (2011) Preparation of anti-zearalenone monoclonal antibodies and development of an indirect competitive ELISA for zearalenone. Microbioloy China 38:1793–1800
Gendloff EH, Casale WL, Ram BP, Tai JH, Pestka JJ, Hart LP (1986) Hapten-protein conjugates prepared by the mixed anhydride method. Cross-reactive antibodies in heterologous antisera. J Immunol Methods 92:15–20
Choi SW, Chang HJ, Lee N, Kim JH, Chun HS (2009) Detection of mycoestrogen zearalenone by a molecularly imprinted polypyrrole-based surface plasmon resonance (SPR) sensor. J Agric Food Chem 57:1113–1118
Liu G, Han Z, Nie D, Yang J, Zhao Z, Zhang J, Li H, Liao Y, Song S, De Saeger S, Wu A (2012) Rapid and sensitive quantitation of zearalenone in food and feed by lateral flow immunoassay. Food Control 27:200–205
Hervas M, Lopez MA, Escarpa A (2011) Integrated electrokinetic magnetic bead-based electrochemical immunoassay on microfluidic chips for reliable control of permitted levels of zearalenone in infant foods. Analyst 136:2131–2138
Gao Y, Yang M, Peng C, Li X, Cai R, Qi Y (2012) Preparation of highly specific anti-zearalenone antibodies by using the cationic protein conjugate and development of an indirect competitive enzyme-linked immunosorbent assay. Analyst 137:229–236
Maragos CM, Appell M (2007) Capillary electrophoresis of the mycotoxin zearalenone using cyclodextrin-enhanced fluorescence. J Chromatogr A 1143:252–257
Panini NV, Salinas E, Messina GA, Raba J (2011) Modified paramagnetic beads in a microfluidic system for the determination of zearalenone in feedstuffs samples. Food Chem 125:791–796
Urraca JL, Benito-Peña E, Pérez-Conde C, Moreno-Bondi MC, Pestka JJ (2005) Analysis of zearalenone in cereal and Swine feed samples using an automated flow-through immunosensor. J Agric Food Chem 53:3388–3344
Wang YK, Zou Q, Sun JH, Wang HA, Yan YX (2012) Detection of zearalenone by chemiluminescence immunoassay in corn samples. Adv Mater Res 550–553:1911–1914
Beloglazova NV, Speranskaya ES, Saeger SD, Hens Z, Abé S, Goryacheva IY (2012) Quantum dot based rapid tests for zearalenone detection. Anal Bioanal Chem 403:3013–3024
Acknowledgments
This work was financially supported by the Chinese National Programs for High Technology Research and Development (2007AA10Z424).
Author information
Authors and Affiliations
Corresponding author
Additional information
Yuan-Kai Wang and Ya-Xian Yan contributed to the work equally and should be regarded as co-first authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 39 kb)
Rights and permissions
About this article
Cite this article
Wang, YK., Yan, YX., Mao, ZW. et al. Highly sensitive electrochemical immunoassay for zearalenone in grain and grain-based food. Microchim Acta 180, 187–193 (2013). https://doi.org/10.1007/s00604-012-0915-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-012-0915-1