
Abstract
We have developed a method, termed solidification of floating organic drop microextraction (SFOME), for the extraction of polybrominated diphenyl ethers (PBDEs) in water and urine samples, this followed by quantification via HPLC. This method requires very small quantities of organic solvent consumption. It is based on exposing a floating solidified drop of an organic solvent on the surface of aqueous solution in a sealed vial. The organic drop is easily collected with a spatula, molten (at ambient temperature), and then submitted to HPLC. Experimental parameters including extraction solvent and its volume, disperser solvent and its volume, extraction time, ionic strength, stirring speed and extraction temperature were optimized. The enrichment factors of analytes are in the range from 921 to 1,462, and acceptable extraction recoveries (92%–118%) are obtained. The dynamic linear range for five PBDE congeners is in the range of 0.5–75 μg.L−1 and from 5 to 500 μg.L−1 for BDE 209. The correlation coefficients range from 0.9960 to 0.9999. The limits of detection (at S/N = 3) for PBDE congeners vary between 0.01 and 0.04 μg.L−1. This method has been successfully applied to detecting PBDEs in two environmental waters and in human urine.

Under optimized conditions, the enrichment factors of PBDEs by solidification of floating organic drop microextraction were from 921 to 1,462, and extraction recoveries (92%–118%) were obtained. The correlation coefficients ranged from 0.9960 to 0.9999. The limits of detection (at S/N = 3) for PBDE congeners varied between 0.01 and 0.04 μg.L−1.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polybrominated diphenyl ethers (PBDEs) are man-made chemicals widely used as flame retardants in a wide variety of paints, plastics, textiles, electronic components, etc. [1–3]. They can escape from the surface of manufactured products and release to the environment. Several epidemiological studies have shown PBDEs to pose health risks [4, 5] such as endocrine disruption, adverse neurobehavioral effects to act as reproductive toxicants and probable carcinogens. Furthermore, toxicological test showed that PBDE congeners might be enriched in liver and thyroid [6, 7]. Thus, some commercial mixtures of PBDEs (penta and octa formulations) have been banned in Europe due to their persistence and potential environmental and human health hazard [8]. However, the major PBDE congeners, such as BDE 47, BDE 99, BDE 100, BDE 154, BDE 183 and BDE 209, are ubiquitous in the environment, and rapidly increasing levels have been frequently detected in waters [9–12], sediments [13, 14], marine organism [15, 16] and food [17, 18].
The direct determination of PBDEs is complicated due to their trace level in complex sample matrices, and thus sample cleanup and pretreatment were required. The reported pretreatment methods for PBDEs in different matrices include stir bar sorption extraction [19, 20], matrix solid-phase dispersion [21], microwave-assisted extraction [22], solid-phase microextraction [23] and dispersive liquid-liquid microextraction [24]. Among them, the dispersive liquid-liquid microextraction is a novel technique, which has been successfully used for extraction and determination of polychlorinated biphenyl [25], polycyclic aromatic hydrocarbons [26], organophosphorus pesticides [27], and some kinds of metal and non-metal in water [28, 29]. The simple and fast microextraction is based on the use of an appropriate extraction and dispersive solvent. The hydrophobic solutes are enriched in the extraction solvent which is dispersed into the bulk aqueous solution. After centrifugation, determination of the analytes in the settled phase can be performed by conventional analytical techniques. However, the extraction solvent in the method is limited. The higher density than water is required for the extraction solvent, the widely used solvents are chlorobenzene, chloroform, tetrachloromethane and carbondisulfide, all of them are toxic and environment-unfriendly [30]. Considering the related problems, another simple, quick and inexpensive microextraction preparation method, i.e., solidification of floating organic drop microextraction (SFOME), has been developed for extraction of analytes from water samples. This technique is based on distribution of analytes between microliter volume of the extraction solvent (floated on the surface of the aqueous sample) and aqueous sample matrix [31].
In SFOME procedures, the suitable organic level at microliter level was floated on the surface of aqueous solution located in a glass vial. The organic solvent must have melting point near room temperature (in the range of 10–30 °C). The aqueous phase was stirred for a selected time [32]. Maximum sensitivity and precision were obtained by stirring the sampling solution until equilibrium was obtained. Then, the sample vial was transferred into a cold water bath for 5 min in order to solidify the organic solvent. The solidified solvent was transferred into a small conical vial by a spatula and melted immediately at ambient temperature. Microliter-level organic solvent was subjected to chromatographic instrument for analysis. This method cannot only avoid using toxic solvent to achieve the idea of green chemistry but also an easy technique compared with other traditional methods. In addition, the experiment time can be shortened from 48 h (Soxhlet extraction) to about 30 min to make the work efficiently [33].
In this study, we used SFOME combined with HPLC to extract six PBDE congeners from water and urine samples, and the main parameters influencing the process were optimized. To the best of authors’ knowledge, this study may be the first report describing the application of SFOME as a preconcentration technique for the analysis of PBDEs from human urine samples.
Experimental
Reagent and standards
2,4,4′-tribrominated diphenyl ether (BDE 28), 2,2′,4,4′-tetrabrominated diphenyl ether (BDE 47), 2,2′,4,4′,5-pentabrominated diphenyl ether (BDE 99), 2,2′,4,4′,5,6′-hexabrominated diphenyl ether (BDE 154) and 2,2′,3,4,4′,5′,6-heptabrominated diphenyl ether (BDE 183) were purchased from J&K, Shanghai, China (www.jkchemical.com). 1,1′,2,2′,3,3′,4,4′,5,5′-decabromodi-phenyl ether (BDE 209) was obtained from Sigma–Aldrich, Shanghai, China (www.sigmaaldrich.com). Each PBDE congener was dissolved in acetonitrile to prepare a 50 mg L−1 stock solution. All the working standard solutions were prepared by serial dilutions of the stock solution with ultra Milli-Q water (www.millipore.com) prior to analysis. The HPLC-grade acetonitrile and methanol were purchased from Merck Company (www.merck.com.cn). 1-undecanol, 1-dodecanol, 2-dodecanol, bromohexadecane and 1,10-dichlorodecane were obtained from Acros J&K, Shanghai, China (www.jkchemical.com).
Water samples were collected from the region far away from e-waste sites (Taizhou, China) and stored in amber bottles at 4 °C until analysis. Urine samples were collected from healthy individuals and stored in polytetrafluoroethylene flasks at −20 °C until analysis. Prior to the SFOME procedures, water samples were filtered through 0.45 μm membrane to remove particulate matter, while urine samples were removed protein by methanol and then filtered through 0.45 μm membrane.
Instrumentation
PBDEs were analyzed by an Agilent 1200 HPLC equipped with a manual injector and variable wavelength detector (VWD). A Zorbax Eclipse XDB-C18 column (150 mm × 4.6 mm, 5 μm particle size) was used and all injections were performed manually with 20.0-μL sample loop. The operating conditions were as follows: mobile phase, methanol-water, 95:5 (v/v); flow rate, 1.0 mL min−1; column temperature, 25 ± 1 °C and the wavelength of detection, 226 nm.
Stirring of the solution was carried out by a model HJ-6A magnetic heater-stirrer with 8 mm × 4 mm string bar, purchased from Jiangsu Jintan Medical Instrument Factory, Jiangsu, China (www.jt-jinyid.com). A 40 mm × 60 mm bottle placed on the heater-stirrer was employed to control temperature of the samples.
Extraction procedure
An aqueous or protein-removed urine solution, containing the mixed PBDE congeners at the concentration of 1 μg L−1 respect to five congeners and BDE 209 at the concentration of 10 μg L−1, was used in the optimization studies. Forty milliliters of the standard solution were transferred into 43 mL vial and microliter volume of 2-dodecanol was placed on the surface of the solution by a 25-μL syringe. The vial was sealed and transferred into the water bath and the stirrer was turned on. When the desired extraction time elapsed, the sample vial was transferred into an ice breaker and the organic solvent was solidified after 10 min. Then, the solidified solvent was transferred into the conical vial and melted quickly. Finally, 5.0 μL of the extractant was injected into the HPLC for quantification. Figure 1 shows schematic diagram of the SFOME apparatus.

Schematic diagram of the SFOME apparatus
Results and discussion
Selection of extraction solvent
An appropriate extraction solvent should meet the following requirements: (1) lower density than water, (2) low volatility and low water solubility, (3) good chromatographic behavior, (4) high extraction capability of analytes and (5) melting point near the room temperature (10–30 °C) [31]. According to the previous considerations, 1-undecanol (melting point, 13–15 °C), 1-dodecanol (22–24 °C), 2-dodecanol (17–18 °C), bromohexadecane (17–18 °C) and 1,10-dichlorodecane(14–16 °C) were investigated in this study. For bromohexadecane, its hydrophobicity was so strong that it can’t be dissolved in the common organic solvent, so it is not suitable for HPLC analysis. The extraction efficiency and chromatographic performance of 1-undecanol, 1-dodecanol and 1,10-dichlorodecane were very poor . 2-dodecanowas found to give the best extraction efficiency, while its chromatographic peak was easily distinguished from analytes of interest. Also, because of its stability, low vapor pressure and low water solubility at the extraction conditions, 2-dodecanol was thus selected as the extraction solvent in this experiment.
Effect of extraction solvent volume
The volume of extraction solvent was another important factor in extraction efficiency of analytes. The effect of extraction solvent volume on extraction efficiency was evaluated by using different volumes of 2-dodecanol (15.0, 20.0, 25.0, 30.0 and 35.0 μL) in this experiment. The relationship between volume of extraction solvent and peak areas is shown in Fig. 2. At larger extraction solvent volume, the settled phase volume increased too, which led to the decreasing of peak area. Thus, the volume of extraction solvent was selected to be 25 μL in this study.

Effect of volume of extraction solvent. Conditions: stirring rate, 900 rpm; sample solution temperature, 60 °C; sample volume, 40 mL; extraction time, 25 min and without salt addition
Effect of sample volume
The effect of sample volume on analytical performance was studied in the range of 10–43 mL (Fig. 3). The results showed that by increasing of sample volume, the peak areas also increased from 10 to 40 mL. However, with the further increase of volumes (>40 mL), the sample vial became unstable and resulted in decreasing of extraction efficiency. Based on LLE equations, rate of the analytes transported into microdrop is directly related to the interfacial area between two liquid phases and inversely related to the organic-phase volume. Thus, by increasing the drop volume, the effect of the interfacial area predominates and the analytical signals are increased. With further increasing of the microdrop volume, the effect of the solvent volume was predominated and the analytical signals are decreased [31]. Therefore, the volume of 40 mL was chosen as the optimal sample volume.

Effect of sample volume. Conditions: stirring rate, 900 rpm; sample solution temperature, 60 °C; extraction solvent volume, 25 μL; extraction time, 25 min and without salt addition
Effect of sample solution temperature, stirring rate, extraction time, ionic strength
The effects of sample solution temperature, stirring rate, extraction time, ionic strength on extraction were also studied. After optimization, at 60 °C sample solution temperature, at 900 rpm stirring rate, 25 min of extraction time, and without salt addition were applied in this experiment. More details about the optimization can be found in the supplementary material.
Analytical performance
For the purpose of quantitative analysis, the calibration curve was obtained under the optimized SFOME-HPLC conditions. The precision of the method was evaluated by carrying out five independent measurements of the studied compounds at 5 μg L−1. Several factors, including linearity ranges, correlation coefficients (R2), and limits of detection were evaluated. As listed in Table 1, the linearity ranges were 0.5–75 μg L−1 for BDE 28, and BDE 47, 0.5–50 μg L−1 for BDE 99, BDE 154 and BDE 183, and 5–500 μg L−1 for BDE 209, respectively. The correlation coefficients (R 2) ranged from 0.9960 to 0.9999. The limits of detection (at S/N = 3) were in the range of 0.01–0.04 μg L−1.
As reported by Zanjani et al. (2007), preconcentration factors (PF) were calculated based on the following equation:
First, an extraction was carried out from a spiked water or urine sample (Caq.ini = 10 μg L−1) by 25 μL 2-dodecanol followed by 5.0 μL injection of the extractant into HPLC. Then a series of standard solutions were prepared in 2-dodecanol as the extraction solution. Finally, by plotting the relative peak area versus concentration for each congener in the standard solutions, the concentrations of PBDE congeners in the organic phase (Co.f) were calculated. From these data, percent extraction (PE) of each PBDE congener extracted into the organic solvent was calculated, using the following equation [31]:
The results are summarized in Tables 1 and 2. The PF values ranged from 921 to 1,462, and the PE values varied between 46% and 74%. The previous results demonstrated that SFOME technique had a high preconcentration capacity for PBDE congeners, which increased about 5–20 times compared with those (in the range of 50–200) obtained by technique of dispersive liquid-liquid microextraction [34].
Real sample analysis
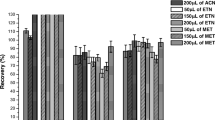
The SFOME technique was applied for the determination of PBDEs in two water and urine samples to clarify applicability and reliability of this method. The results showed that the residues of PBDE congeners were all at below detectable level (<0.01 μg L−1) in water and urine sample with exception that BDE 99 and BDE 183 were detected to be 1.0 and 2.1 μg L−1, respectively, in river water 1#. The previous samples were then spiked with standards of PBDE congeners at different levels to assess the matrix effect. The spiking recoveries of the target PBDEs in the real samples are summarized in Table 3. In the spiked levels of 5, 10 and 50 μg L−1, the relative recoveries ranged from 81% to 118% in water samples. In the spiked level of 5 and 50 μg L−1, the relative recoveries were in the range of 92%–118% in human urine samples. The previous results demonstrated that the SFOME method could be used in trace PBDE analysis for the environmental water and human urine samples. Figure 4 shows the chromatograms attained from blank (a) and spiked urine samples (b) at the concentration level of 50 μg L−1 for PBDEs according to SFOME method.

Chromatograms attained from the blank (a) and spiked urine samples (b) at the concentration level of 50 μg L−1 for PBDEs according to SFOME method
Comparison with other methods
Extraction and determination of PBDEs in water samples by the SFOME-HPLC method was compared with other methods. As summarized in Table 3, lower LODs and RSDs in SFOME-HPLC were obtained. Moreover, the present technique provided higher PF in comparison with SDME-HPLC and DLLME-HPLC. In general, it was a very simple, rapid, inexpensive and environmentally friendly technology.
Conclusions
This study illustrated the successful application of SFOME technique that allows the separation and preconcentration of PBDEs present at low concentration levels in environmental water and human urine samples. In comparison with other extraction methods, the present method only requires less-toxic solvent and also has good relative recovery. Additionally, a detection limit at ng.L−1 level was achieved due to the higher PF values (921–1,462) compared with dispersive liquid-liquid microextraction (50–500) and single-drop microextraction (20–200) techniques. The main drawback of this pretreatment technique is the limitation on selection of extraction solvent because of overlapping of solvent peak with some analyte peaks. However, many solvents are available that have suitable melting points. Overall, it is an environmentally friendly technique and most importantly, we can shorten the experiment from 48 h (soxhlet extraction) to less than 30 min to make the work efficient. Therefore, this method has a great potential in the routine multi-residual analysis of PBDEs at trace levels in environmental water and human urine samples.
References
Dufault C, Poles G, Driscoll LL (2005) Brief postnatal PBDE exposure alters learning and the cholinergic modulation of attention in rats. Toxicol Sci 88:172–180
Kodavanti PR, Ward TR, Ludewig G, Robertson LW, Birnbaum LS (2005) Polybrominated diphenyl ether (PBDE) effects in rat neuronal cultures: 14C-PBDE accumulation, biological effects, and structure-activity relationships. Toxicol Sci 88:181–192
Viberg H, Fredriksson A, Eriksson P (2005) Deranged spontaneous behaviour and decrease in cholinergic muscarinic receptors in hippocampus in the adult rat, after neonatal exposure to the brominated flame-retardant, 2,2′,4,4′,5-pentabromodiphenyl ether (PBDE 99). Environ Toxicol Pharmacol 20:283–288
Fernie KJ, Mayne G, Shutt JL, Pekarik C, Grasman KA, Letcher RJ, Drouillard K (2005) Evidence of immunomodulation in nestling American kestrels (Falco sparverius) exposed to environmentally relevant PBDEs. Environ Pollut 138:485–493
Nakari T, Pessala P (2005) In vitro estrogenicity of polybrominated flame retardants. Aquat Toxicol 74:272–279
Tseng LH, Li MH, Tsai SS, Lee CW, Pan MH, Yao WJ, Hsu PC (2008) Developmental exposure to decabromodiphenyl ether (PBDE 209): effects on thyroid hormone and hepatic enzyme activity in male mouse offspring. Chemosphere 70:640–647
Costa LG, Giordano G (2007) Developmental neurotoxicity of polybrominated diphenyl ether (PBDE) flame retardants. Neurotoxicology 28:1047–1067
KemmLein S, Herzke D, Law RJ (2003) BFR-governmental testing programme. Environ Int 29:781–792
Hyötyläinen T, Hartonen K (2002) Determination of brominated flame retardants in environmental samples. Trends Anal Chem 21:13–30
Cheung KC, Poon BH, Lan CY, Wong MH (2003) Assessment of metal and nutrient concentrations in river water and sediment collected from the cities in the Pearl River Delta, South China. Chemosphere 52:1431–1440
Zheng GJ, Martin M, Richardson BJ, Yu H, Liu Y, Zhou C, Li J, Hu G, Lam MH, Lam PK (2004) Concentrations of polybrominated diphenyl ethers (PBDEs) in Pearl River Delta sediments. Mar Pollut Bull 49:514–524
Li Y, Hu J, Liu X, Fu L, Zhang X, Wang X (2008) Dispersive liquid-liquid microextraction followed by reversed phase HPLC for the determination of decabrominated diphenyl ether in natural water. J Sep Sci 31:2371–2376
Pan J, Yang YL, Xu Q, Chen DZ, Xi DL (2007) PCBs, PCNs and PBDEs in sediments and mussels from Qingdao coastal sea in the frame of current circulations and influence of sewage sludge. Chemosphere 66:1971–1982
Cai ZW, Jiang GB (2006) Determination of polybrominated diphenyl ethers in soil from e-waste recycling site. Talanta 70:88–90
Päpke O, Fürst P, Herrmann T (2004) Determination of polybrominated diphenylethers (PBDEs) in biological tissues with special emphasis on QC/QA measures. Talanta 63:1203–1211
Akutsu K, Obana H, Okihashi M, Kitagawa M, Nakazawa H, Matsuki Y, Makino T, Oda H, Hori S (2001) GC/MS analysis of polybrominated diphenyl ethers in fish collected from the inland sea of Seto, Japan. Chemosphere 44:1325–1333
She J, Holden A, Sharp M, Tanner M, Williams-Derry C, Hooper K (2007) Polybrominated diphenyl ethers (PBDEs) and polychlorinated biphenyls (PCBs) in breast milk from the Pacific Northwest. Chemosphere 67:S307–S317
Pirard C, De Pauw E (2007) Absorption, disposition and excretion of polybrominated diphenyl ethers (PBDEs) in chicken. Chemosphere 66:320–325
Serôdio P, Cabral MS, Nogueira JM (2007) Use of experimental design in the optimization of stir bar sorptive extraction for the determination of polybrominated diphenyl ethers in environmental matrices. J Chrom A 1141:259–270
Prieto A, Zuloaga O, Usobiaga A, Etxebarria N, Fernández LA (2008) Use of experimental design in the optimisation of stir bar sorptive extraction followed by thermal desorption for the determination of brominated flame retardants in water samples. Anal Bioanal Chem 390:739–748
Martínez A, Ramil M, Montes R, Hernanz D, Rubí E, Rodríguez I, Cela Torrijos R (2004) Development of a matrix solid-phase dispersion method for the screening of polybrominated diphenyl ethers and polychlorinated biphenyls in biota samples using gas chromatography with electron-capture detection. J Chrom A 1072:83–91
Wang JX, Jiang DQ, Gu ZY, Yan XP (2006) Multiwalled carbon nanotubes coated fibers for solid-phase microextraction of polybrominated diphenyl ethers in water and milk samples before gas chromatography with electron-capture detection. J Chrom A 1137:8–14
Bayen S, Lee HK, Obbard JP (2004) Determination of polybrominated diphenyl ethers in marine biological tissues using microwave-assisted extraction. J Chrom A 1035:291–294
Li YY, Wei GH, Hu J, Liu XJ, Zhao XN, Wang XD (2008) Dispersive liquid–liquid microextraction followed by reversed phase-high performance liquid chromatography for the determination of polybrominated diphenyl ethers at trace levels in landfill leachate and environmental water samples. Anal Chim Acta 615:96–103
Rezaei F, Bidari A, Birjandi AP, Milani Hosseini MR, Assadi Y (2008) Development of a dispersive liquid-liquid microextraction method for the determination of polychlorinated biphenyls in water. J Hazard Mater 158:621–627
Rezaee M, Assadi Y, Milani Hosseini MR, Aghaee E, Ahmadi F, Berijani S (2006) Determination of organic compounds in water using dispersive liquid-liquid microextraction. J Chrom A 1116:1–9
Berijani S, Assadi Y, Anbia M, Milani Hosseini MR, Aghaee E (2006) Dispersive liquid-liquid microextraction combined with gas chromatography-flame photometric detection: very simple, rapid and sensitive method for the determination of organophosphorus pesticides in water. J Chrom A 1123:1–9
Naseri MT, Hosseini MR, Assadi Y, Kiani A (2008) Rapid determination of lead in water samples by dispersive liquid-liquid microextraction coupled with electrothermal atomic absorption spectrometry. Talanta 75:56–62
Shokoufi N, Shemirani F, Assadi Y (2007) Fiber optic-linear array detection spectrophoto- metry in combination with dispersive liquid-liquid microextraction for simultaneous preconcentration and determination of palladium and cobalt. Anal Chim Acta 597:349–356
Lv LL, Xu H, Song D, Cui Y, Hu S, Zhang G (2010) Analysis of volatile aldehyde biomarkers in human blood by derivatization and dispersive liquid-liquid microextraction based on solidification of floating organic droplet method by high performance liquid chromatography. J Chrom A 1217:2365–2370
Khalili Zanjani MR, Yamini Y, Shariati S, Jönsson JA (2007) A new liquid-phase microextraction method based on solidification of floating organic drop. Anal Chim Acta 585:286–293
Khalili-Zanjani MR, Yamini Y, Yazdanfar N, Shariati S (2008) Extraction and determination of organophosphorus pesticides in water samples by a new liquid phase microextraction gas chromatography-flame photometric detection. Anal Chim Acta 606:202–208
Chang C, Huang S (2010) Determination of the sterod hormone levels in water samples by dispersive liquid-liquid microextraction with solidification of a floating organic drop followed by high performance liquid chromatography. Anal Chim Acta 662:39–43
Li YY, Wei GH, Wang XD (2007) Determination of decabromodiphenyl ether in water samples by single-drop microextraction and RP-HPLC. J Sep Sci 30:2698–2702
Cortazar E, Zuloaga O, Sanz J, Raposo JC, Etxebarria N, Fernández LA (2002) Multisimplex optimization of the solid-phase microextraction gas chromatographic-mass spectrometric determination of polycyclic aromatic hydrocarbons, polychlorinated biphenyls and phthalates from water samples. J Chrom A 978:165–175
Acknowledgements
This work was jointly funded by “Xinmiao Talent” Project of Zhejiang Province (2010R413061), Public Benefit Project of Zhejiang Province (2011C23112, 2011C37006), National Natural Science Foundation of China (21077079), Key Project of Wenzhou City (20082780125, Z090921421), and International Cooperation Project of Wenzhou Science and Technology Bureau (H20090079, H20100053, H20100054).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 4286 kb)
Rights and permissions
About this article
Cite this article
Liu, H., Zhang, M., Wang, X. et al. Extraction and determination of polybrominated diphenyl ethers in water and urine samples using solidified floating organic drop microextraction along with high performance liquid chromatography. Microchim Acta 176, 303–309 (2012). https://doi.org/10.1007/s00604-011-0713-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-011-0713-1