
Abstract
We describe a simple and rapid method for the ultrasound-assisted microextraction of antimony using the solidified floating organic drop method. The effects of pH, type and volume of the extractant, time of sonication, amount of chelating agent, type and amount of surfactant were investigated and optimized. Bromopyrogollol red is acting as the chelating agent. Antimony(III) ion was extracted into finely dispersed droplets of undecanol after ion-pair formation with the water soluble chelator and the cationic detergent benzyldimethyltetradecylammonium chloride. Flame atomic absorption spectrometry was used for the detection. The resulting calibration is linear in the concentration range from 4.0 to 900 ng mL-1 of Sb(III) with a correlation coefficient of 0.9981. The enrichment factor is 67, the detection limit is 0.62 ng mL-1, and the relative standard deviation is ± 3.6% (at 100 ng mL-1; for n = 10). The method was successfully applied to the determination of antimony in water samples.

Antimony and many of its compounds are toxic and can damage the kidneys and the liver, causing death in a few days. Concentration of this element is very low in nature and hence their determination required sensitive analytical techniques. One such technique is an ultrasound assisted emulsification microextraction procedure.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Sampling and sample pretreatment typically account for most of the time required for analysis and the quality of these steps largely determines the success of an analysis from a complex matrix. Since most analytical instruments cannot directly handle complex matrices, we need a new method for extracting, isolating and concentrating the analytes of interest [1, 2]. Traditional methods for sample preparations, including liquid-liquid extraction (LLE), solvent extraction, distillation, precipitation, and absorption are tedious and time-consuming. They are also multistage operations, and to some extent difficult to automate [3]. Furthermore, they usually require large amounts of high purity organic solvents, which are potentially toxic and expensive [4]. Consequently, the current trend is towards simplification and miniaturization of the sample-preparation steps which decrease the use of such solvents [5]. A simple, inexpensive liquid-phase microextraction (LPME) was introduced recently [6]. Three new methodologies have arisen based on this method: single-drop microextraction (SDME), hollow fibre liquid-phase microextraction (HF-LPME) and dispersive liquid–liquid microextraction (DLLME) [7–9]. Pedersen-Bjergaard and Rasmussen [10], introduced an alternative concept for LPME based on the use of single, low cost, disposable, porous, hollow fibers, typically made of polypropylene [11]. In 2006, Assadi et al. [12] developed a dispersive liquid-liquid microextraction (DLLME) technique. This method is based on the use of an appropriate extractant, it uses microlitres of an organic solvent with high density and a disperser organic solvent with high miscibility in both extractant and aqueous phase. The extraction procedure begins when the mixture of extractant and disperser solvent is rapidly injected into the sample solution and a cloudy solution is formed [13, 14]. In 2007, Khalili-Zanjani et al. introduced a simple and inexpensive liquid-phase microextraction method based on the solidification of a floating organic droplet (LPME-SFO) [15] in which an extractant with a lower density than that of water, low toxicity, and melting point near room temperature (in the range of 10–30 °C) was used. In 2008, Leong and Huang [16] reported a novel variation of SFODME. Instead of maintaining one droplet of extractant in the sample, a dispersion of fine droplets was formed by injecting a mixed solution of the extractant and dispersive solvent. This produces a vast contact area between the extractant and the sample, leading to faster mass transfer and better extraction times. A new method using solidified floating organic drop microextraction, based on ultrasound-dispersion was developed [17]. Hence, ultrasonic waves, play the role of the stirrer bar and dispersive solvent in conventional SFODME and DLLME, respectively. Some researchers prefer to use ultrasonic waves not only to accelerate the formation of a cloudy dispersive extraction mixture but also for transfer of the analyte between two immiscible phases, thus resulting in a lower extraction time [18–22]. Antimony and many of its compounds are toxic. The effects of antimony poisoning are similar to arsenic poisoning [1, 23], inhalation of antimony dust is harmful and in certain cases may be fatal. In small doses, antimony causes headaches, dizziness, and depression and larger doses such as prolonged skin contact, may cause dermatitis. It can also damage the kidneys and the liver, causing violent and frequent vomiting, and death in a few days [24]. Antimony and its compounds are considered to be priority interest pollutants by the Environmental Protection Agency of the United States [25], with a maximum allowable contaminant level of 6 μg L−1 in drinking water. This metal occurs mainly in two oxidation states. The toxicity of Sb (III) is 10–20 times higher than that of Sb (V), and their oxides can cause several types of cancer [26, 27]. Since the concentrations of this element in water are very low, sensitive analytical techniques are required. Inductively coupled plasma mass spectrometry (ICP-MS) [28, 29], inductively coupled plasma atomic emission spectrometry (ICP-AES) [29, 30], hydride generation atomic absorption spectrometry (HG-AAS) [31] and electrothermal atomic absorption spectrometry (ETAAS) [32, 33] have been recommended for this purpose. A number of separation and determination procedures involving hydride generation [34], liquid–liquid extraction [35], solid phase extraction (SPE) [36, 37], coprecipitation [38], dispersive liquid–liquid microextraction (DLLME) [39], Headspace single-drop microextraction [40], hydride generation fluorescence spectrometry [41] and cloud point extraction (CPE) [42] have been proposed. In this study, the possibility of Sb (III) enrichment by ultrasound- assisted emulsification microextraction (USAEME) is considered. Bromopyrogollol red is selected as the chelating reagent and a new microextraction method combined with flame atomic absorption spectrometry (FAAS) is developed for separation, enrichment and determination of Sb(III) in aqueous samples.
Experimental
Apparatus
A Varian SpectrAA 220 atomic absorption spectrometer (Varian, Australia, http://www.varianinc.com) equipped with deuterium background correction was employed with an antimony hollow-cathode lamp operating at 10 mA, as a source of radiation. Measurements were carried out in the integrated absorbance mode at 217.6 nm, using a spectral bandwidth of 0.2 nm. The acetylene and air flows were 1.0 and 3.5 L min–1, respectively. A Metrohm 827 pH meter (Herisau, Switzerland, http://www.metrohm.com) was used for pH measurements. An IEC HN-S centrifuge (New York, USA, http://www.medwow.com) was used to accelerate the phase separation. Fine droplets of the organic solvent were produced by a Sonorex RK255 ultrasonic water bath (Bandelin, Germany, http://www.ultraschall-anlagen.de).
Chemicals and reagents
A stock standard solution of Sb (ΙΙΙ) at a concentration of 1000.0 μg mL−1 was purchased from Merck (Darmstadt, Germany, http://www.merck.de). Working standard solutions were prepared daily by serial dilutions of the stock solution with double distilled water immediately prior to analysis. A 0.01% w/v solution of the chelating agent bromopyrogollol red (BPR) (Geel, Belgium http://www.acros.com) was prepared by dissolving an appropriate amount of BPR in double distilled water. 1-Undecanol, 1-hexadecanethiol, 2-undecanone were obtained from Merck. Methanol, acetone, ethanol and DMF as diluents solvents were purchased from Merck. Benzyldimethyltetradecyl-ammonium chloride-dihydrate (BDTA) (Merck) was used as a surfactant for ion pair formation. Buffer solutions were prepared from acetic acid and sodium acetate for pH 3.1-6, sodium dihydrogen phosphate and disodium hydrogen phosphate for pH 6–8. The solutions of alkali metal salts and various metal salts were used to study the interference of anions and cations, respectively. The laboratory glassware was kept in a 1.0 mol L−1 HCl solution overnight and rinsed thoroughly with double distilled water.
USAEME procedure
The schematic procedure of the USAEME is shown in Fig. 1. 20.0 mL of the aqueous solution containing 100.0 ng mL−1 of antimony (III), 1.0 mL BDTA 1% (w/v) and 1.0 mL BPR 0.01% (w/v) was placed in a screw cap glass tube after the pH of the sample was adjusted to 6.2 by using 0.1 mol L−1 sodium dihydrogen phosphate and disodium hydrogen phosphate buffer. In this step, after reaction of Sb (III) ions with BPR, 45 μL of 1-undecanol were added and the sample was sonicated for 2 min. A cloudy solution resulting from the dispersion of fine 1-undecanol droplets in the aqueous solution was formed in the test tube. This cloudy solution was centrifuged for 8 min at 1250 rpm leading to the aggregation of 1-undecanol as a floating drop on the surface of the solution. In this step the Sb (III) complex was extracted into 1-undecanol and then the sample vial was transferred into an ice bath where the organic solvent was solidified after 10 min. The solidified solvent was then transferred into a conical vial where it immediately melted and was diluted to 300 μL with ethanol. It was then transferred to the FAAS for determination of Sb (III).

Schematic procedure of UA-SFODME. a Injection of extractant into aqueous sample; b ultrasound-assisted formation of emulsion; c collection of solidified extractant after centrifugation and cooling the sample; d dilution
Results and discussion
In order to demonstrate the capability of USAEME for separation and preconcentration of Sb (III) ion, BPR (which is widely used for determination of metal ions) was selected as the chelating agent. 1-Undecanol was also used as extracting solvent because of its low volatility, low water solubility and melting point (13–15 °C) near room temperature. Furthermore, to obtain a high enrichment factor, different parameters affecting the complex formation, extraction and analysis processes were optimized in a one-at-a-time approach. Also, the extraction recovery (ER) and enrichment factor were calculated according to Eqs. 1 and 2
where V and C are the volume and concentration and the suffixes o and aq indicate organic and aqueous phase, respectively [17].
Effect of pH
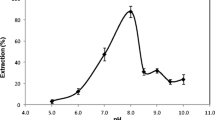
It is obvious that the pH of the solution plays a unique role on metal-chelate formation. In the present work, the effect of pH on the extraction recovery of Sb (III) ion was studied within the pH range of 3.5–8.5. The results illustrated in Fig. 2 reveal that the recovery is nearly constant in the pH range of 5.7–6.5. The progressive decrease in extraction of Sb (III) ion at low pHs is due to the competition of hydrogen ion with other analytes for reaction with BPR. For pHs greater than 6.5, decrease in extraction is due to the precipitation of antimony as antimony hydroxide. In subsequent studies, the pH of the solution was adjusted to ~ 6.2 using sodium dihydrogen phosphate and disodium hydrogen phosphate buffer.

Effect of pH on the extraction recovery of antimony. Conditions: Sb (III), 100.0 ng mL-1; BDTA 1% (w/v), 1 mL; BPR 0.01% (w/v), 1 mL; sonication time, 2 min; extraction solvent, 1- undecanol, 45.0 μL; centrifugation time, 8 min
Effect of BPR amount
The effects of 0.01% (w/v) BPR volume on the extraction recovery of analytes were evaluated in the range of 0.2–1.600 mL. The recovery reaches a maximum at 1.0 mL and is then independent of further increase in the volume of BPR at a concentration greater than 1.0 mL. A volume of 1.0 mL was selected as optimum for further studies.
Selection of extracting solvent
The extraction solvent must satisfy certain conditions (low volatility, lower density than water and low water solubility) in order to be stable during the extraction period, extract analytes well, easily separate from watery phase and have a melting point near room temperature (in the range of 10–30 °C). Several extracting solvents, including 1-undecanol (mp 13–15 °C), 1-hexadecanethiol (mp 18–20 °C) and 2-undecanone (mp 11–13 °C) were investigated. 1-Undecanol was found to give the best extraction efficiency. In addition, it has sensitivity, stability, lower price, low water solubility and low vapor pressure. Thus, 1-undecanol was selected as the extracting solvent. In fact, with 2-undecanone as the extraction solvent, the dispersed drop could not be aggregated completely after centrifugation. This maybe because of its solubility in water (0.00179 g/100 mL at 25 °C)
Effect of the volume of extracting solvent
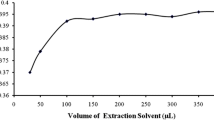
The volume of extraction solvent is a crucial parameter and has an important effect on the extraction efficiency and also determines enrichment performance to study the effect of extraction solvents. Different volumes of 1-undecanol (35, 40, 45, 50, 55, 60 and 75 μL) were used as extraction solvents in identical USAEME procedures. As can be seen, in Fig 3, the recovery of extraction stays constant with the increase of 1-undecanol volume in the range of 35–45 μL, and then decreases with further increases in volume. Therefore, 45 μL of 1-undecanol was selected for subsequent experiments.

Effect of extraction solvent volume on the extraction recovery of antimony. Conditions were the same as Fig. 2 except extraction solvent volume
Effect of sonication time
Sonication time plays an important role in the USAEME procedure. Enough time will make the extracting solvent disperse finely into the aqueous solution and result in an excellent cloudy solution and hence a high enrichment factor. The effect of ultrasonic agitation time was evaluated in the range of 1–7 min. Extraction recoveries were 95%, 99%, 95%, 93%, 87%, 85%, and 80% as the sonication time increased from 1 min to 7 min respectively. The corresponding results show that recovery increased up to 2 min and then decreased. The extracting solvent disperses more finely into the aqueous solution and the two phases cannot be distinguished after centrifuging. Hence, 2 min was used for the dispersive procedure.
Effect of centrifugation time
Centrifugation was necessary to separate phases in the extraction tubes. The effect of centrifugation time on the extraction efficiency was evaluated in the range of 4–12 min at 1250 rpm. It was found that the extraction recovery was best when the solution was centrifuged at 1250 rpm for 8 min and remained constant after that.
Effect of surfactant
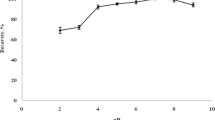
Three kinds of surfactant, Triton X-100, sodium didodecyl sulfate and BDTA, were tested for extraction of antimony complex. The best recovery was obtained using BDTA. The volume of BDTA was investigated in the range of (0.1–1.3 mL). Figure 4 shows that when the volume of BDTA was 1.0 mL, the antimony complex extracts completely.

Effect of surfactant amount on the extraction recovery of antimony. Conditions were the same as Fig. 2 except BDTA amount
Effect of extraction time
The effect of extraction time was examined in the range of 10 s to 30 min. The results showed that the extraction time had no notable effect on the enrichment factor.
Effect of potentially interfering ions
In order to demonstrate the selectivity of the presented method for the separation of antimony ions, the effect of common potentially interfering ions in water samples was investigated and the potential interference was evaluated. Interference ions, in different interference-to-analyte ratios, were added to a solution containing fixed amount of antimony (100.0 ng mL−1) and were subjected to the USAEME procedure. The results of this investigation are given in Table 1. An ion that causes a variation of more than ±5% in absorbance of antimony was consider as potentially interfering ion. As is shown in the table, at the given levels, most ions show no significant interference in the determination of antimony. However, metal ions including Cu2+ and V5+ may interfere in the extraction recovery of antimony.
Figures of merit
The analysis of the USAEME method, including the quantification, reproducibility, limit of detection, enrichment factor, linear range, calibration curve and precision were determined to evaluate method performance. The calibration graph was obtained after the standard series were subjected to the USAEME and then determined by FAAS. The linear regression equation was A = 0.237 C +0.020 (where A is absorbance and C is the concentration (ng mL-1 of antimony), linearity was obtained with antimony concentration in the range of 4.0–900 ng mL-1. The enrichment factor for the presented method was obtained as 66.6 and the limit of detection based on three times the standard deviation of the blank solution measurements (n = 10) divided by the slope of the calibration curve was 0.63 ng mL−1. The relative standard deviation for ten replicate determination of 100.0 ng mL−1 of Sb (III) was ±3.6%.
Application to real and synthetic samples
To demonstrate the performance of the proposed method, it was used to extract and determine antimony concentration in different water samples including tap water (Kerman drinking water), well water (University of Kerman) and mineral water. The antimony concentration in water samples was out of the calibration range. So, for reliability of antimony determination from water samples, specified amounts of antimony were added to the sample and measurements were taken. As may be seen in Table 2, the recoveries of the spiked samples are satisfactory. We also used this method to separate, recover and determine the amount of antimony in synthetic samples. The results, given in Table 3, are in agreement with the certified values. Therefore, the method is quite accurate.
Analysis of antimony in standard alloys
The method was applied to the determination of antimony in Nippon Keikinzoku Kogyo (NKK CRM 916 aluminum alloy). A 0.1 g sample of standard aluminum alloy was first dissolved completely in 6–14 mL of hydrochloric acid by heating on a water-bath at 85 °C and then 1 mL of 30% (v/v) hydrogen peroxide and 5 mL of hydrofluoric acid (to remove interference of aluminum) were added to it. The excess of peroxide was decomposed by heating in the water bath and the solution was cooled, filtered and diluted to 100 mL with distilled water in a standard flask. An aliquot (1–2 mL) of this sample was taken and analyzed by the general procedure. The results are given in Table 4.
Comparison of USAEME with other methods
The results for a comparison of USAEME combined with flame atomic absorption spectrometry with other techniques for separation of antimony is given in Table 5. In comparison with other reported methods, the USAEME- FAAS method shows a comparatively low detection limit (0.63 ng mL-1), a high enrichment factor (66.6), good dynamic range (4.0–900 ng mL-1) in a short extraction time. In addition, because only very small amounts of volatile organic materials were used, our method is safe. The relative standard deviation was ±3.6%. All these results indicate that USAEME- FAAS is a reproducible, sensitive, simple, green and low cost technique that can be used for determination of metal ions like antimony from water samples. Therefore, the method is of interest, especially for laboratories doing routine trace metal ion analysis
Conclusion
An ultrasonic instrument was used to accelerate the formation of a cloudy dispersed extraction mixture in the short time of 2 min. In this study, ultrasound- assisted emulsification microextraction coupled with flame atomic absorption spectrometry, has been developed for separation and sensitive determination of antimony in aqueous samples. It has also been shown that the SbBPR--BDTA+ can be extracted into 1-undecanol. This technique, which has no need for a dispersive solvent as in the conventional DLLME, minimizes the sample preparation time and the consumption of organic solvents. The results also indicate that this extraction procedure has outstanding advantages such as simplicity, low cost, high enrichment factor, and rejection of matrix constituents. Future work will be directed at extraction of other metal ions using various ligands and the assessment of the multi-element enrichment capability of the method for ultra trace determination in different matrices.
References
Inen THT, Riekkola ML (2008) Sorbent and liquid-phase microextraction techniques and membrane-assisted extraction in combination with gas chromatographic analysis: a review. Anal Chim Acta 614:27
Yazdi AS, Amiri A (2010) Liquid-phase microextraction. TrAc Trends Anal Chem 29:1
Ying WY, Ying ZG, Yun CQ, Huan ZX, Chun W, Zhi W (2010) Developments in liquid-phase microextraction method based on solidified of floating organic drop. Chin J Anal Chem 38:1517
Mirzaei M, Behzadi M, Abadi NM, Beizaei A (2011) Simultaneous separation/preconcentration of ultra trace heavy metals in industrial wastewaters by dispersive liquid–liquid microextraction based on solidification of floating organic drop prior to determination by graphite furnace atomic absorption spectrometry. J Hazard Mater 186:1743
Pereira FP, Lavilla I, Bendicho C (2009) Miniaturized preconcentration methods based on liquid–liquid extraction and their application in inorganic ultratrace analysis and speciation: a review. Spectrochim Acta B 64:15
Jeannot MA, Przyjazny A, Kokosa JM (2010) Single drop microextraction development, applications and future trends. J Chromatogr A 1217:2336
Bidabadi MS, Dadfarnia S, Shabani AMH (2009) Solidified floating organic drop microextraction (SFODME) for simultaneous separation/preconcentration and determination of cobalt and nickel by graphite furnace atomic absorption spectrometry (GFAAS). J Hazard Mater 166:296
Jeannot MA, Cantwell FF (1996) Solvent microextraction into a single drop. Anal Chem 68:2240
Liu HH, Dasgupta PK (1996) Analytical chemistry in a drop. solvent extraction in a microdrop. Anal Chem 68:1821
Bjergaard SP, Rasmussen KE (1999) Liquid-liquid-liquid microextraction for sample preparation of biological fluids prior to capillary electrophoresis. Anal Chem 71:2656
Payan MR, Lopez MAB, Fernandez-Torres R, Navarro MV, Mochon MC (2009) Hollow fiber-based liquid-phase microextraction (HF-LPME) of ibuprofen followed by FIA-chemiluminescence determination using the acidic permanganate–sulfite system. Talanta 79:915
Rezaee M, Assadi Y, Milani-Hosseini MR, Aghaee E, Ahmaidi F, Berijani S (2006) Determination of organic compounds in water using dispersive liquid–liquid microextraction. J Chromatog A 1116:9
Takahiko B, Fumio K, Susumu N, Katsuroku T (2009) Study of drop coalescence behavior for liquid–liquid extraction operation. Chem Eng Sci 55:5391
Yan H, Wang H, Qin X, Liu B, Du J (2011) Ultrasound-assisted dispersive liquid–liquid microextraction for determination of fluoroquinolones in pharmaceutical wastewater. J Pharmaceut Biomed 54:57
Zanjani MRK, Yamini Y, Shariati S, Jonsson JA (2007) A new liquid-phase microextraction method based on solidified of floating organic drop. Anal Chim Acta 585:293
Leong MI, Huang SD (2008) Dispersive liquid–liquid microextraction method based on solidified of floating organic drop for extraction of organochlorine pesticides in water samples. J Chromatogr A 1211:12
Mohamadi M, Mostafavi A (2010) A novel solidified floating organic drop microextraction based on ultrasound-dispersion for separation and preconcentration of palladium in aqueous samples. Talanta 81:313
Regueiro J, Llompart M, Garcia-Jares C, Garcia-Monteagudo JC, Cela R (2008) Ultrasound-assisted emulsification-microextraction of emergent contaminants and pesticides in environmental waters. J Chromatogr A 1190:27
Mohammadi SZ, Afzali D, Taher MA, Baghelani YM (2010) Determination of trace amounts palladium by atomic absorption spectrometry after ligandless-dispesive liquid-liquid microextraction. Microchim Acta 168:123
Fontana AR, Wuilloud RG, Martinez LD, Altamirano JC (2009) Simple approach based on ultrasound-assisted emulsification-microextraction for determination of polibrominated flame retardants in water samples by gas chromatography–mass spectrometry. J Chromatogr A 1216:147
Mohammadi SZ, Afzali D, Taher MA, Baghelani YM, Karimzadeh L (2011) Ultrasound-assisted emulsification microextraction of trace amounts of Co and Mn ions prior to flame atomic absorption spectrometry. J Braz Chem Soc 22:104
Tajik S, Taher MA (2011) New method for microextraction of ultra trace quantities of gold in real samples using ultrasound-assisted emulsification of solidified floating organic drops. Microchim Acta 173:249
Lide DR (2008) Magnetic susceptibility of the elements and inorganic compounds, in Handbook of chemistry and Physics 81st edition. CRC press, Taylor & Francis Group
Oehme FW (1978) Toxicity of heavy metals in the environment. Dekker, New York
United States Environmental Protection Agency, USEPA, 1 (1979) EP- 440/4-79-029A
Karim MM, Komori Y, Alam M (1997) Subsurface arsenic occurrence and depth of contamination in Bangladesh. J Environ Chem 7:792
Poon R, Chu I, Lacavalier P, Valli V, Foster GWS, Thomas B (1998) Effects of antimony on rats following 90-day exposure via drinking water. Food Chem Toxicol 36:35
Yu CH, Cai QT, Guo ZX, Yang ZG, Khoo SB (2002) Antimony speciation by inductively coupled plasma mass spectrometry using solid phase extraction cartridges. Analyst 127:1385
Partha C, Fisher AS, Henon DN, Hill SJ (2004) Matrix digestion of soil and sediment samples for extraction of lead, cadmium and antimony and their direct determination by inductively coupled plasma-mass spectrometry and atomic emission spectrometry. Microchim Acta 144:277
Feng YL, Narasaki H, Chen HY, Tian LC (1999) Speciation of antimony(III) and antimony (V) using hydride generation inductively coupled plasma atomic emission spectrometry combined with the rate of pre-reduction of antimony. Anal Chim Acta 386:304
Cabon JY, Madec CL (2004) Determination of major antimony species in seawater by continuous flow injection hydride generation atomic absorption spectrometry. Anal Chim Acta 504:215
Ojeda CB, Rojas FS, Pavon JMC, Martin LT (2005) Use of 1,5-bis (di-2-pyridyl) methylene thiocarbohydrazide immobilized on silica gel for automated preconcentration and selective determination of antimony (III) by flowinjection electrothermal atomic absorption spectrometry. Anal Bioanal Chem 382:329
Safilov T, Angelov N, Jacimovic R, Stibilj V (2005) Determination of trace elements in arsenic and antimony minerals by atomic absorption spectrometry and k0-instrumental neutron activation analysis after removal of As and Sb. Microchim Acta 149:229
Kumar AR, Riyazuddin P (2007) Non-chromatographic hydride generation atomic spectrometric techniques for the speciation analysis of arsenic, antimony, selenium, and tellurium in water samples: a review. Int J Environ An Ch 87:500
Calleguntinas MBDL, Madrid Y, Camara C (1992) Speciation of antimony by atomic absorption spectrometry. Applicability to selective determination of antimony (III) and antimony (V) in liquid samples and of bioavailable antimony in sediments and soil samples. Microchim Acta 109:155
Perenyi KZ, Jankovics P, Sugar E, Lasztity A (2008) Solid phase chelating extraction and separation of inorganic antimony species in pharmaceutical and water samples for graphite furnace atomic absorption spectrometry. Spectrochim Acta B 63:449
Zhang L, Morita Y, Sakuragawa A, Isozaki A (2007) Inorganic speciation of As (III, V), Se (IV, VI) and Sb (III, V) in natural water with GF-AAS using solid phase extraction technology. Talanta 72:729
Sun YC, Yang JY (1999) Simultaneous determination of arsenic (III, V), selenium (IV, VI), and antimony (III, V) in natural water by coprecipitation and neutron activation analysis. Anal Chim Acta 395:300
Rivas RE, Lopez-Garcia I, Hernandez-Cordoba M (2009) Speciation of very low amounts of arsenic and antimony in waters using dispersive liquid–liquid microextraction and electrothermal atomic absorption spectrometry. Spectrochim Acta B 64:329
Pena-Pereira F, Lavilla I, Bendichob C (2009) Headspace single-drop microextraction with in situ stibine generation for the determination of antimony (III) and total antimony by electrothermal-atomic absorption spectrometry. Microchim Acta 164:77
Wu H, Wang X, Liu B, Liu Y, Li S, Lu J, Tian J, Zhao W, Yang Z (2011) Simultaneous speciation of inorganic arsenic and antimony in water samples by hydride generation-double channel atomic fluorescence spectrometry with on-line solid-phase extraction using single-walled carbon nanotubes micro-column. Spectrochim Acta B 66:80
Jiang X, Wen S, Xiang G (2010) Cloud point extraction combined with electrothermal atomic absorption spectrometry for the speciation of antimony(III) and antimony(V) in food packaging materials. J Hazard Mater 175:150
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Iraji, A., Afzali, D., Mostafavi, A. et al. Ultrasound- assisted emulsification microextraction for separation of trace amounts of antimony prior to FAAS determination. Microchim Acta 176, 185–192 (2012). https://doi.org/10.1007/s00604-011-0706-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-011-0706-0