
Abstract
We report on a method for trace analysis of the narcotic drugs alfentanil, fentanyl, and sufentanil in plasma and urine. Two–phase hollow fiber liquid–phase microextraction was combined with GC using nitrogen–phosphorus detection. Experimental parameters were optimized to give a viable analytical procedure whose limits of detection range from 8 to 15 ng L−1 (at an S/N of 3). The calibration curves are linear between 0.1 and 50 μg L−1, with squared correlation coefficients (r 2) between 0.9953 and 0.9979. Precision values range from 2.4% to 3.3% (intra–day RSD) and 3.2 to 6.3% (inter–day RSD). The relative recoveries varied from 27.8% to 84.6% (for spiked plasma) and 75 to 85.2% (for spiked urine). The method consumes little solvent, is simple, fast, inexpensive, and well suitable for the analysis of complicated matrices.

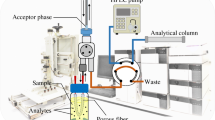
Schematic diagram of two phase hollow fiber liquid-phase microextraction (HF-LPME) combined with GC and nitrogen–phosphorus detection for trace analysis of the narcotic drugs alfentanil, fentanyl, and sufentanil in plasma and urine.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Fentanyl, alfentanil and sufentanil are synthetic opioid and known in the drug scene have been shown to be much more potent than morphine derivatives. They are widely used for the purpose of neuroleptic analgesia and sedation during preoperative, induction, maintenance, and postoperative surgical periods. Sufentanil is the most potent of the series and is about 2,000 times more potent than morphine [1]. Alfentanil and fentanyl exhibit about 30 and 100 times the potency of morphine, respectively [2, 3]. Due to their efficacy at low dosages, blood and urine concentration of these drugs are below the limit of detection of most assay methods. Therefore, the detection of very low concentration (at ng L−1 level) of the compounds from analgesic doses is important.
Several analytical methods have been applied to determine fentanyl, sufentanil and alfentanil in biological samples. Radioimmunoassay and enzyme–linked immunosorbent assay have been used for detection at ng L−1 level. However, lack of selectivity and poor precision are among the main disadvantages of these techniques [4–7]. GC–NPD [8], HPLC–UV [9], GC–MS [10–13] and LC–MS/MS [14] have also been developed. However, due to low concentration of the analytes in urine and plasma, a sample preparation and cleanup step must be performed before determination. Liquid–liquid extraction (LLE) and solid phase extraction (SPE) combined with HPLC [15], GC [16] or GC–MS [17, 18] have been used successfully to determine narcotic drugs in biological fluids. Although these techniques provide adequate analyte enrichment, they are time consuming and require large volumes of toxic solvents.
In recent years, different microextraction techniques such as solid–phase microextraction (SPME) and liquid–phase microextraction (LPME) have been introduced as alternatives to LLE and SPE [19, 20]. SPME and LPME have been successfully applied for the analysis of a wide range of analytes in various matrices [21–26]. Hollow fiber liquid–phase microextraction (HF–LPME) has originally been introduced by Pedersen–Bjergaard and Rasmussen [27] in 1999 with its basic principles having been clearly described in several reviews [28–30]. In one of the possible configurations, (two phase HF–LPME), analytes are extracted from an aqueous solution through pores of a porous hollow fiber of polypropylene into a small volume of an organic solvent, which can be directly analyzed via GC. The method is almost solvent free, simple, and fast. Of course, some drawbacks, such as air bubble formation on the membrane surface, poor reproducibility because of manual cutting and sealing of the hollow fiber have been reported for HF-LPME [28–30].
Despite advantages and wide uses of HF–LPME, It has not been used for analysis of fentanyl, sufentanil and alfentanil. The aim of this work is develop a method for simultaneous determination of three narcotic drugs (fentanyl, sufentanil and alfentanil) at sub ppb level in plasma and urine samples by HF–LPME and GC–NPD.
Experimental
Chemicals and reagents
Fentanyl citrate, alfentanil hydrochloride, and sufentanil citrate were obtained from Tofigh Daru (Tehran, Iran, www.tofighdarou.tapic.co.ir). Stock standard solutions of individual compounds were prepared in methanol (alfentanil, fentanyl, and sufentanil with concentrations of 860, 760 and 940 mg L−1, respectively). An intermediary standard solution at a concentration of 100 mg L−1 was prepared by diluting the standard solution in methanol. More diluted working solutions were prepared daily by diluting the intermediary standard solution with pure water. Water samples were prepared by spiking pure water with analytes at known concentrations to study the extraction performance under different conditions. The stock solution of internal standard (I.S.) was prepared by dissolving 9.9 mg of trioctylamine in 10 mL methanol. All solutions were stored at 4 °C prior to use. HPLC–grade methanol, water and acetone were purchased from Caledon Laboratories (Georgetown, Ont., Canada, www.caledonlabs.com). Diphenyl ether, n-decane, 1,2,4-trimethylbenzene, n-octanol, and hexyl acetate were supplied from Merck (Darmstadt, Germany, www.merck.com).
The pH of water samples was adjusted with sodium hydroxide (Merck). Other reagents and solvents were also purchased from Merck.
Instrumentation
A SP–3420 gas chromatograph equipped with a split/splitless injector and a nitrogen phosphorus detector (BFRL, Beijing, China, www.bfrl.com.cn) was used. Nitrogen (99.999%) was used as a carrier and make–up gas. The carrier and make–up gas flow rate were set at 2 and 30 mL min−1, respectively. The components were separated using a DB–35MS, 30 m × 0.25 mm, capillary column with a 0.15 μm stationary phase thickness (J&W Scientific, Folsom, CA, USA, www.jandw.com). The injector and detector temperatures were set at 260 and 300 °C, respectively. The column was initially maintained at 110 °C, held for 1 min; subsequently, the temperature was increased to 280 °C at a rate of 30 °C min−1 (3 min hold) and was increased to 300 °C (25 °C min−1) and held for 3 min. Hydrogen and air flow for NPD were adjusted at 5.5 and 175 mL min−1, respectively. The NPD bead was turned on 5 min after injection.
HF–LPME procedure
Q3/2 Accurel polypropylene hollow fiber membrane (600 μm inner diameter, 200 μm wall thickness and 0.2 μm pore size) was purchased from Membrana GmbH (Wuppertal, Germany, www.membrana.com). Hollow fibers were cut into 1 cm pieces, washed with acetone in ultrasonic bath to remove any contaminants and dried. A 3-mL sample solution containing NaOH (0.005 M) spiked with an appropriate amount of studied compounds was introduced in a 4 mL glass vial. The vial was placed on a digital magnetic stirrer (MR 3000D, Heidolph, Germany, www.heidolph.com). The needle tip of a 25-μL microsyringe was inserted into the hollow fiber and the lumen of the fiber was filled with hexyl acetate containing internal standard. The open end of the fiber was sealed with the aid of a tweezers. The fiber was then immersed in hexyl acetate for about 30 s to impregnate pores of the fiber and then rinsed with water by being inserted in water for 10 s in order to remove excessive organic solvent. Extraction of the analytes from the sample solution to the solvent within the fiber was performed under solution stirring at 1,000 rpm for 15 min at room temperature. After extraction, the fiber was taken out, the end was cut and the organic solvent was withdrawn into the syringe. The hollow fiber was detached and discarded. The extract was injected into the GC in splitless mode.
Real samples
Drug-free urine sample was obtained from a healthy person while drug-free plasma was obtained from blood transfusion organization (Isfahan, Iran) and thawed at room temperature before use. To eliminate particulate matters, the samples were filtered through a 0.45-μm nylon membrane filter (Millipore, Bedford, MA, USA, www.millipore.com). The NaOH concentration of the sample was adjusted at 0.005 M and extracted using the HF–LPME procedure. In the case of plasma samples, in order to reduce the matrix effects, the sample was diluted with pure water (1:1) before extraction.
Results and discussion
Optimization of HF–LPME conditions
To obtain the best sensitivities, different parameters affecting HF–LPME were investigated. These parameters include organic extraction solvent, NaOH concentration in sample solution, ionic strength, stirring rate, extraction time, and temperature. All determinations were based on the relative peak area of analytes to the internal standard, from the average of three replicate measurements.
Selection of the extraction solvent
Careful attention should be paid to the selection of the organic solvent immobilized in the pores of the hollow fiber, which is a significant factor for HF–LPME. The range of solvents suitable for use with hollow fiber membranes is limited as the solvent should be easily immobilized in the fiber pores, immiscible with water, have low volatility to prevent solvent loss during extraction, and be compatible with the GC system. Several different organic solvents, such as n-decane, diphenyl ether, hexyl acetate, n-octanol, and 1,2,4-trimethylbenzene were investigated in this study. To evaluate extraction capability of the solvents, HF–LPME was performed using different solvents for 15 min. Water samples were spiked with the analytes at 0.3 mg L−1 concentration level. The organic solvents contained trioctylamine (I.S.) at 5 mg L−1. The results showed that hexyl acetate gave better extraction efficiency (Fig. 1).

Effect of solvent type on HF–LPME. Extraction conditions: concentration of analytes: 0.3 mg L−1; sample volume: 3.0 mL; NaOH concentration as donor solution: 0.005 M; without salt addition; stirring rate: 1,000 rpm; extraction time: 15 min; room temperature
Although n-octanol is almost the most used extraction solvent in solvent microextraction techniques [24, 26, 28], it has lower extraction efficiency than that of the hexyl acetate utilized in this work. Our previous studies [31, 32] also confirm suitability of hexyl acetate as an extraction solvent compared to other organic solvents used in SDME and LPME methods. At the same time, it has very low toxicity compared to other common organic solvents such as octanol, toluene, dodecane and benzene. Consequently, hexyl acetate was selected as the extraction solvent in further experiments.
NaOH concentration of sample solution
For the aqueous solution containing acidic or basic drugs, the donor solution pH is adjusted to neutralize the analytes and thereby affect their water solubility and extractability into the organic solvent. Drugs used in this study are basic; therefore, the pH of the aqueous solution should be higher than their pKa. To study the effect of sample basicity, the NaOH concentration was varied between 0.0005 and 0.05 M. Extraction efficiency increased with increasing NaOH concentration up to 0.005 M. The decrease in the response at higher NaOH concentration (>0.005 M) could be attributed to the increasing ionic strength of the sample solution by increasing NaOH concentration [9]. Based on the results, 0.005 M of NaOH was selected for the subsequent analysis.
Salt addition
To investigate the influence of sample ionic strength on the affinities of analytes toward the organic solvent, different amounts of NaCl (0–35% (w/v)) were added into the solution. As can be seen in Fig. 2, increasing salt concentration led to a decrease in the extraction efficiency of fentanyl and sufentanil. For alfentanil, the extraction efficiency appeared to reach a maximum at a NaCl concentration of 0.2 g mL−1 and subsequently decreased with increasing salt content. Addition of salt has a mutual effect [33, 34]. First, it might change the physical properties of the Nernst diffusion film and reduce the diffusion rate of the target compounds into the extraction solvent. Second, it can improve the extraction efficiency of the analytes due to salting out effect. Therefore, depending on the dominating factor, the extraction efficiency would either increase or decrease. Based on the above experimental data, all the following experiments were carried out without adding salt.

Effect of ionic strength on HF–LPME. Extraction conditions: concentration of analytes: 0.3 mg L−1; sample volume: 3.0 mL; organic solvent: hexyl acetate; NaOH concentration as donor solution: 0.005 M; stirring rate: 1,000 rpm; extraction time: 15 min; room temperature
Stirring rate
Stirring the solution facilitates the mass transfer process and reduces the time required to reach thermodynamic equilibrium and thereby, increases the extraction efficiency. Therefore, in order to obtain the best sensitivity, stirring rates between 600 and 1,200 rpm were evaluated. The relative peak area of the analytes increases with increasing the stirring rate up to 1,200 rpm (the highest speed that could be achieved by the magnetic stirrer). In order to achieve better precision, the stirring rate of 1,000 rpm was selected for subsequent extractions.
Sample temperature
Temperature has a significant influence on both kinetics and thermodynamics of the extraction process. High temperature increases the diffusion rate of the analytes and reduces the time required for reaching equilibrium. On the other hand, with increasing temperature, the distribution constant of analytes to the organic phase are decreased. In addition, depletion of the organic solvent may occur at a higher temperature and thus extraction efficiency would be diminished [32, 35]. To evaluate the effect of extraction temperature, the sample temperature varied in the range 25–55 °C. The highest relative responses for all the compounds were obtained at room temperature (25 °C).
Extraction time
HF–LPME is based on the equilibrium of analytes between the organic solvent in the hollow fiber, and the sample solution. To extract the maximum amount of analytes, the effect of extraction time in the yield of the method was studied. Therefore, extractions were carried out at 5, 10, 15, and 20 min. The relative peak area increased with the increasing of extraction time up to 15 min. After 15 min, peak intensity was decreased. This is probably due to solvent loss and air bubble formation at the fiber surface [36]. The creation of air bubbles on the surface of the hollow fiber reduces the transport rate of analytes into organic solvent and decreases the amount of analytes extracted. According to the results, 15 min was selected for extraction time in subsequent studies.
Method evaluation
The optimum HF–LPME parameters were selected as: 3.0 mL sample solution, 1 cm porous hollow fiber immobilized with hexyl acetate as the organic solvent, 0.005 M NaOH as donor solution, 1,000 rpm stirring rate, 15 min extraction time, without salt addition at room temperature. To evaluate the practical applicability of the HF–LPME technique, analytical performance data of the method (i.e. linearity, precision, enrichment factor, and detection limit) were investigated under the optimal conditions. The results are shown in Table 1.
Enrichment factor
HF–LPME is a non–exhaustive extraction method which is not expected to obtain high enrichment factor. The EF was calculated using the following equation:
Where CAP and CDP are the final and initial concentrations of the analyte in the extraction solvent after HF-LPME and donor phase, respectively. The enrichment factors were obtained by three replicate extractions of water samples spiked at 5 μg L−1 of the analytes. Under the optimal conditions, the results indicate that EFs were between 35 and 64.
Linearity, precision, and detection limit
The linearity of calibration curves was evaluated by spiking deionized water over a concentration range of 0.1–50 μg L−1 for all compounds. All of the analytes show good linearity with squared correlation coefficient (r 2) ranging from 0.9953 to 0.9979. The intra- and inter-day precision of the developed method was studied in terms of the relative standard deviations (RSDs). The intra-day precision was studied for five replicate extractions of spiked deionized water samples containing 1 μg L−1 of the analytes. The inter-day precision of the method was carried out by performing three consecutive extractions each day over a period of three working days. Intra–day RSDs ranged between 2.4% and 3.3%, and inter–day RSDs ranged between 3.2% and 6.3%. The limits of detection (LODs), being calculated with S/N = 3 on the basis of peak-to-peak noise, were in the range of 8–15 ng L−1.
Biological samples
In order to investigate the capability of the method in analyzing real samples and the effect of sample matrix on the determination of narcotic drugs, the present LPME method was used for the analysis of biological samples. Analysis was performed by four-point standard addition technique with the plasma and urine samples being spiked with a mixture of the analytes at a concentration level between 0.2 and 2 μg L−1. As shown in Table 2, a good linear relationship between spiked amounts and the relative peak area of all the analytes was observed. To evaluate the relative recovery (RR%) and RSDs, the samples were extracted after being spiked with 4 μg L−1 of the studied compounds. The relative recoveries of the analytes in the samples were obtained from the following equation:
Where, A1, A2 and A3 are peak areas of the analyte in spiked sample extract, unspiked sample extract and spiked distilled water extract, respectively. The relative recoveries obtained by the method (Table 2) were higher than 75% for urine sample with a relative standard deviation lower than 9.8%. This demonstrates that the influence of the matrix was not significant for the analysis of the analytes in urine samples. For alfentanil determination in plasma sample, RR% is 84.6. However, the relative recovery obtained for fentanyl and sufentanil are relatively low (40.6 and 27.8, respectively). The low recovery obtained for these two compounds may be attributed to the high degree of protein binding [29, 37]. To release the drugs from the protein, sample pH must be adjusted at alkaline region to deionize the analytes. However, for some drugs the addition of methanol (5–50% (v/v)) was required to eliminate drug-protein interaction and obtain high recoveries [29, 37]. In LPME method the addition of methanol into the donor solution could lead to very low recovery due to the increase in water solubility of the analytes and dissolution of organic solvent. Despite the low extraction recovery of fentanyl and sufentanil from plasma samples, because of high sensitivity of the NPD, the method detection limits are still low enough to determine the analytes in plasma samples. On the other hand, acceptable RSDs and good correlation coefficients obtained by the method for plasma samples (lower than 11.8%) ensure a precise analysis.
The chromatograms obtained for non-spiked and spiked plasma and urine samples at 4 and 2 μg L−1 of the analytes, respectively, are shown in Figs. 3 and 4. None of the analytes were found in the samples. The raise in the baseline is mainly due to establishing thermal equilibrium of the bead after turning on. In addition, bleeding of the column is increased as the temperature increases.

Obtained chromatograms of the three narcotic drugs after HF–LPME of blank and spiked plasma sample at 4 μg L−1

Obtained chromatograms of the three narcotic drugs after HF–LPME of blank and spiked urine sample at 2 μg L−1
Conclusions
A HF–LPME–GC–NPD method was developed for the extraction and determination of three narcotic drugs in human plasma and urine without any additional sample pretreatment step such as protein precipitation, centrifugation, etc. Matrix components of the real samples do not interfere with quantitation of the analytes. The method exhibited good linearity, precision, and detection limit. At the same time, it was fast, simple, sensitive, inexpensive, and allowed sample extraction and clean-up to be done in a single step. The present LPME sample preparation method offers good potential as a feasible alternative to LLE, SPE and SPME techniques for the extraction of the analytes in plasma and urine samples. Compared with the above mentioned extraction methods (Table 3) for the preparation of urine and plasma samples, the presented HF-LPME method enjoyed numerous merits such as simplicity, low cost, easy/fast operation and low detection limits. Despite the fact that recoveries obtained for plasma sample were relatively low, the sensitivity of the method was good enough for the determination of the analytes.
References
Clotz MA, Nahata MC (1991) Clinical uses of fentanyl, sufentanil and alfentanil. Clin Pharm 10:581–593
Scholz J, Steinfath M, Schulz M (1996) Clinical pharmacokinetics of alfentanil, fentanyl and sufentanil–an update. Clin Pharmacokinet 31:275–292
Valaer AK, Huber T, Andurkar SV, Clark CR, De Ruiter J (1997) Development of a gas chromatographic–mass spectrometric drug screening method for the N–dealkylated metabolites of fentanyl, sufentanil, and alfentanil. J Chromatogr Sci 35:461–466
Fryirsa B, Woodhouse A, Huang JL, Dawson M, Mather LE (1997) Determination of subnanogram concentrations of fentanyl in plasma by gas chromatography–mass spectrometry: comparison with standard radioimmunoassay. J Chromatogr B 688:79–85
Ruangyuttikarn W, Law MY, Rollins DE, Moody DE (1990) Detection of fentanyl and its analogs by enzyme–linked immunosorbent assay. J Anal Toxicol 14:160–164
Dotsikas Y, Loukas YL, Siafaka I (2002) Determination of umbilical cord and maternal plasma concentrations of fentanyl by using novel spectrophotometric and chemiluminescence enzyme immunoassays. Anal Chim Acta 459:177–185
Dotsikas Y, Loukas YL (2004) Employment of 4–(1–imidazolyl)phenol as a luminol signal enhancer in a competitive–type chemiluminescence immunoassay and its comparison with the conventional antigen–horseradish peroxidase conjugate–based assay. Anal Chim Acta 509:103–109
Raikos N, Theodoridis G, Alexiadou E, Gika H, Argiriadou H, Parlapani H, Tsoukali H (2009) Analysis of anesthetics and analgesics in human urine by headspace SPME and GC. J Sep Sci 32:1018–1026
Ebrahimzadeh H, Yamini Y, Gholizade A, Sedighi A, Kasraee S (2008) Determination of fentanyl in biological and water samples using single–drop liquid–liquid–liquid microextraction coupled with high–performance liquid chromatography. Anal Chim Acta 626:193–199
Wang C, Li E, Xu G, Wang H, Gong Y, Li P, Liu S, He Y (2009) Determination of fentanyl in human breath by solid–phase microextraction and gas chromatography–mass spectrometry. Microchem J 91:149–152
Gupta PK, Manral L, Ganesan K, Dubey DK (2007) Use of single–drop microextraction for determination of fentanyl in water samples. Anal Bioanal Chem 388:579–583
Van Nimmen NFJ, Poels KLC, Veulemans HAF (2004) Highly sensitive gas chromatographic—mass spectrometric screening method for the determination of picogram levels of fentanyl, sufentanil and alfentanil and their major metabolites in urine of opioid exposed workers. J Chromatogr B 804:375–387
Dufresne C, Favetta P, Gonin R, Bureau J, Guitton J (2002) Simultaneous determination of fentanyl and midazolam in plasma using direct solid–phase microextraction before gas chromatography–mass spectrometry analysis. Anal Lett 35:1575–1590
Huynh NH, Tyrefors N, Ekman L, Johansson M (2005) Determination of fentanyl in human plasma and fentanyl and norfentanyl in human urine using LC–MS/MS. J Pharm Biomed Anal 37:1095–1100
Ghazi-Khansari M, Zendehdel R, Pirali-Hamedani M, Amini M (2006) Determination of morphine in the plasma of addicts in using Zeolite Y extraction following high–performance liquid chromatography. Clin Chim Acta 364:235–238
Soriano C, Munoz-Guerra J, Carreras D, Rodriguez C, Rodriguez AF, Cortes R (1996) Automated analysis of drugs in urine. J Chromatogr B 687:183–187
Meadway C, George S, Braithwaite R (2002) A rapid GC–MS method for the determination of dihydrocodeine, codeine, norcodeine, morphine, normorphine and 6–MAM in urine. Forensic Sci Int 127:136–141
Kudo K, Ishida T, Hara K, Kashimura S, Tsuji A, Ikeda N (2007) Simultaneous determination of 13 amphetamine related drugs in human whole blood using an enhanced polymer column and gas chromatography–mass spectrometry. J Chromatogr B 855:115–120
Prosen H, Zupancíicí-Kralj L (1999) Solid–phase microextraction. Trends Anal Chem 18:272–282
Sarafraz-Yazdi A, Amiri A (2010) Liquid–phase microextraction. Trends Anal Chem 29:1–14
Barri T, Jönsson J-Å (2008) Advances and developments in membrane extraction for gas chromatography: Techniques and applications. J Chromatogr A 1186:16–38
Chen Y, Guo Z, Wang X, Qiu C (2008) Sample preparation. J Chromatogr A 1184:191–219
Pawliszyn J (1999) Applications of solid phase microextraction. Royal Society of Chemistry, Cambridge
Pedersen-Bjergaard S, Rasmussen KE (2008) Liquid–phase microextraction with porous hollow fibers, a miniaturized and highly flexible format for liquid–liquid extraction. J Chromatogr A 1184:132–142
Ridgway K, Lalljie SPD, Smith RM (2007) Sample preparation techniques for the determination of trace residues and contaminants in foods. J Chromatogr A 1153:36–53
Xu L, Basheer C, Lee HK (2007) Developments in single–drop microextraction. J Chromatogr A 1152:184–192
Pedersen-Bjergaard S, Rasmussen KE (1999) Liquid–liquid–liquid microextraction for sample preparation of biological fluids prior to capillary electrophoresis. Anal Chem 71:2650–2656
Psillakis E, Kalogerakis N (2003) Developments in liquid–phase microextraction. Trends Anal Chem 22:565–574
Rasmussen KE, Pedersen-Bjergaard S (2004) Developments in hollow fiber–based, liquid–phase microextraction. Trends Anal Chem 23:1–10
Lee J, Lee HK, Rasmussen KE, Pedersen-Bjergaard S (2008) Environmental and bioanalytical applications of hollow fiber membrane liquid–phase microextraction: a review. Anal Chim Acta 624:253–268
Saraji M, Farajmand B (2008) Application of single–drop microextraction combined with in–microvial derivatization for determination of acidic herbicides in water samples by gas chromatography–mass spectrometry. J Chromatogr A 1178:17–23
Saraji M, Mousavi F (2010) Use of hollow fiber–based liquid–liquid–liquid microextraction and high–performance liquid chromatography–diode array detection for the determination of phenolic acids in fruit juices. Food Chem 123:1310–1317
Xiao Q, Hu B, Yu C, Xia L, Jiang Z (2006) Optimization of a single–drop microextraction procedure for the determination of organophosphorus pesticides in water and fruit juice with gas chromatography–flame photometric detection. Talanta 69:848–855
De Jager LS, Andrews AR (2001) Development of a screening method for cocaine and cocaine metabolites in urine using solvent microextraction in conjunction with gas chromatography. J Chromatogr A 911:97–105
Li G, Zhang L, Zhang Z (2008) Determination of polychlorinated biphenyls in water using dynamic hollow fiber liquid–phase microextraction and gas chromatography–mass spectrometry. J Chromatogr A 1204:119–122
Xia L, Hu B, Wu Y (2007) Hollow fiber–based liquid–liquid–liquid microextraction combined with high–performance liquid chromatography for the speciation of organomercury. J Chromatogr A 1173:44–51
Pedersen-Bjergaard S, Rasmussen KE (2005) Bioanalysis of drugs by liquid–phase microextraction coupled to separation techniques. J Chromatogr B 817:3–12
Bagheri H, Es-haghi A, Khalilian F, Rouini MR (2007) Determination of fentanyl in human plasma by head–space solid–phase microextraction and gas chromatography–mass spectrometry. J Pharm Biomed Anal 43:1763–1768
Acknowledgments
The authors acknowledge Research Council of Isfahan University of Technology (IUT) and Center of Excellence in Sensor and Green Chemistry for the financial support of this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Saraji, M., Boroujeni, M.K. Analysis of narcotic drugs in biological samples using hollow fiber liquid–phase microextraction and gas chromatography with nitrogen phosphorus detection. Microchim Acta 174, 159–166 (2011). https://doi.org/10.1007/s00604-011-0612-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-011-0612-5