
Abstract
A method is presented for magnetic solid phase extraction (MSPE) of tetracyclines in milk samples. The method involves the extraction and clean-up by silica based magnetic support dispersion on non-pretreated milk samples, followed by the magnetic isolation and desorption of the analytes by acidified methanol. The tetracyclines eluted from the magnetic support were determined simultaneously by flow injection analysis with spectrophotometric detection. Under optimal conditions, the linear range of the calibration curve ranges from 0.03 to 0.60 mg L−1, with a limit of detection of 10 μg L−1. Recoveries were determined for milk spiked at levels from 0.15 to 0.60 mg L−1. Average recoveries ranged from 91.0 to 97.0%, with a precision of <5.0% in all cases. The method was validated by comparing the results with those obtained by MSPE-HPLC and SPE-HPLC. No significant differences were observed (p < 0.05)
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Tetracyclines (TCs) are broad-spectrum antibiotic agents extensively used to control bacterial infections in humans and animals. They exhibit activity against infections caused by Gram-positive and Gram-negative bacteria. TCs are given to food producing animals as therapeutic agents or as growth promoters with exception of European Union, were only therapeutic use is allowed [1]. In dairy cows significant percentage of the administrated TCs is excreted in milk, therefore TCs may be present in marketed products and cause harmful effects to consumers, including possible allergic reactions, liver damage, yellowing of teeth, and gastrointestinal disturbance. In addition, trace amounts of antibiotic compounds in milk favor the development of antibiotic-resistant bacteria. In order to protect humans from the exposure to these drug residues in milk the EU has established a maximum residue limit (MRL) of 0.1 mg L−1 for tetracycline (TC), oxytetracicline (OT) and chlortetracycline (CT) [1]. The U.S. Food and Drug Administration (FDA) has established the tolerance of 0.3 mg L−1 for the combined residues of TC, OTC and CT [2].
With the arguments stated above, the determination of residual TCs in animal products is consecutively important. Microbiological assays and immunoassays are routinely used as screening methods to detect tetracycline residues in food despite their disadvantages such as: poor sensitivity and selectivity, semi-quantitative measurements of residues detected, and false positive results. HPLC and capillary electrophoresis are also applied for TCs confirmation in food and animal origin products [3, 4]. On the other hand, flow injection analysis (FIA) is a well established automated technique with numerous applications in quantitative chemical analysis; it is a simple and inexpensive technique [5]. Compared to separations techniques such as chromatography, FIA offers increased sampling rate, lower reagents consumption, and similar precision and accuracy. These advantages have led to a growing interest in food analysis and quality control applications [6, 7].
Sample preparation processes including both clean-up and preconcentration are indispensable during drug residues determination in food samples. Liquid-liquid extraction [8], solid phase extraction [9], matrix solid phase dispersion [10], ion exchange [11], and molecularly imprinted polymers [12] have been applied during TCs isolation from complex matrices. Magnetically assisted chemical separation, which uses nano- or micron-sized particles, has attracted great deal of research interest on sample preparation since the past decade. Magnetic supports are commonly prepared by organic/inorganic polymerization in presence of magnetite. Magnetic solid phase extraction (MSPE) is a current separation procedure based on the use of magnetic adsorbents. In this procedure, the magnetic support is dispersed in the sample and after the suspension is formed, the support (with the analytes adsorbed) is collected by an external magnetic field without additional centrifugation or filtration. The analytes are consequently eluted from the recovered adsorbent and the extract is analyzed. The main advantages of using magnetically assisted separation in comparison to conventional pretreatment methods are its easier, faster, environmental friendly and cheaper handling [13].
Silica-coated magnetite particles are perhaps one of the most ideal options for MSPE since silica shell has reliable stability in aqueous media and a large surface area. TCs are highly adsorbed on silica-based materials, thus these materials are good candidates for TCs preconcentration in milk samples when using magnetic solid phase extraction [14].
In this work, a MSPE for TCs preconcentration, from milk samples, based on magnetic silica coated materials is developed. The effect of several parameters on TCs isolation was evaluated employing a Taguchi parameter design. In order to increase the sampling rate, the analysis was carried out by FIA with spectrophotometric detection which allowed the simultaneous determination of TCs. The method was validated comparing the results obtained with those provided by HPLC.
Experimental
Reagents and materials
All solutions were prepared by dissolving the respective analytical grade reagent in deionized water with a resistivity not less than 18.0 MΩ cm provided by a Milli-Q system (Millipore, Bedford, MA, USA). Boric acid, methanol, tetracycline hydrochloride (95%), oxytetracycline dehydrate (95%), chlortetracycline hydrochloride (95%), 4-aminoantipyrine (AAP) and potassium hexacyanoferrate (II)) were purchased from Sigma (St Louis, MO, USA, www.sigmaaldrich.com). EDTA sodium salt, sodium hydroxide and hydrochloric acid were obtained from J.T. Baker (Phillipsburg, NJ, USA, www.waters.com).
Separate standard solutions of each TC compound were prepared at a concentration of 1 mg mL−1 by dissolving the pure substances in deionized water. Working solutions were prepared daily by dilution of the standard stock solution in deionized water. Calibration standards were prepared at concentrations of 0.10–0.60 mg L−1, as the sum of TCs. AAP carrier solution (1.8% w/v) was prepared by dissolving 4-aminoantipyrine in borate buffer solution (1.0 mol L−1, pH 10.0). The oxidizing reagent solution was hexacyanoferrate (III) (0.8% w/v) dissolved in borate buffer solution. Solid phase extraction was applied using Sep-Pack Vac C18 cartridges (Waters, Milford, MA, USA). A total of 22 milk samples were bought from different supermarkets in Pachuca, Mexico.
Instrumentation
The FIA system [15] (Fig. 1) consists of a four-channel Gilson Minipuls 2 peristaltic pump fitted with propulsion tubes of the same brand used to propel sample and carrier solutions. A sample volume of 400 μl was injected into the AAP solution (CS), using a Rheodyne 5020 four-way rotary valve, the phenolic ring present in TCs reacts with AAP in a reaction coil (R1, 30 cm × 0.8 mm i.d.). The coupled compound produced was joined and mixed with the hexacyanoferrate (III) solution (OS) in a reaction coil (R2, 90 cm × 0.8 mm i.d). The quinone-imide compound generated was detected at 540 nm in an UV-visible spectrophotometer (Lambda 40, Perkin-Elmer, www.perkinelmer.com) with a Hellma 178.712QS 18 μL flow-through cell. The flow rate of each individual channel was 2.0 mL min−1. The different components of the FIA manifold were connected using Omnifit Teflon tubing (0.8 mm internal diameter) and Gilson connectors.

Flow injection manifold used to determine TCs. S, sample; CS, Coupling solution (AAP, 1.8% w/v); OS, oxidizing solution (K3Fe(CN)6, 0.8% w/v); PP, peristaltic pump; V, injection valve; R1 and R2, reaction coil; D, detector; W, waste
Chromatography experiments were done using a PerkinElemer Series 200 liquid chromatography system (PerkinElemer MA, USA) equipped with a UV-Vis detector at 360 nm and a manual injector connected to a 20 μL external loop. Chromatographic separation was achieved with a Spheri-5 ODS column (5 μm; 240 × 4.6 mm) from PerkinElmer. The mobile phase was 0.01 mol L−1 aqueous oxalic acid: methanol:acetonitrile (64:18:18 v/v) at a flow rate of 1.0 mL min−1 [9].
Preparation of the magnetic support
Magnetite (Fe3O4) was prepared by coprecipitation method, as follows: 0.03 mol FeSO4.7H2O were dissolved in 100 mL of water. The mixture was stirred vigorously while the temperature was increased to 60°C. A stream of air was bubbled in the mixture whilst a NaOH solution (2 mol L−1) was added to adjust pH value to 10. After 1.0 h, the magnetic precipitates were isolated from the solvent by a permanent magnet and washed several times with deionized water. The magnetite previously synthesized (1.5 g) was added to a flask containing 5.95 g of tetramethoxysilane solubilized in 24 mL of a solution that contained: Triton X-100 (2.0% w/v), CTAB (0.2% w/v), methanol (12.5% v/v) and 200 μL f NH3 (28%, v/v). The mixture was heated and refluxed at 120°C for 1 h with stirring. The chemical characterization of this support has been described in a previous work [16].
Nitrogen sorption experiments were performed at 77°K on a Micrometrics ASAP 2020 analyzer. The samples were outgassed at 100°C for 10 h before the measurement. Brunauer-Emmett-Teller (BET) surface area was calculated from adsorption data in the relative pressure range from 0.01 to 0.2. The pore size distribution curves were obtained from the adsorption branches using Barrett-Joyner-Halenda (BJH) method. Particle size distribution was determined using a Beckman Coulter LS 13320 light scattering particle size distribution analyzer with a diode laser of wavelength 750 nm. The sample was dispersed in a Triton X-100 (0.2% w/v) solution at 25°C in order to disperse the solid particles in the mixture.
Sample preparation
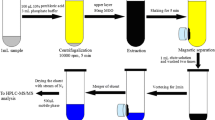
In the MSPE procedure using silanized magnetite, 0.3 g of the magnetic support were added into 50 mL of milk. The mixture was stirred for 20 min to form a homogeneous dispersion. After standing for 5 min, the magnetic support (containing the analytes) was separated from the sample using an external magnetic field. The liquid phase was disposed and the magnetic support isolated was flushed with 10 mL volumes of EDTA (1 × 10−3 mol L−1) and deionized water. TCs were eluted from the magnetic support with 3 × 1.5 mL of acetic acid (1 × 10−3 M) in methanol and the eluates were combined. A 0.5 mL aliquot of this methanolic solution was transferred into a 10 mL calibration flask, treated with 0.5 mL of EDTA 1 × 10−3 mol L−1 and diluted to volume with borate buffer solution and finally injected into the FIA system. In HPLC experiments the methanolic eluate was filtered through a 0.45 μm filter and injected into the chromatography system.
The SPE was performed following the method reported [9] by J.W. Fritz et al. 10 mL of milk samples were mixed with 30 mL of the McIlvine/EDTA solution. The mixture was agitated and centrifuged for 15 min or until the protein precipitated. The precipitate was disposed off and the solution was applied to a SPE cartridge, which was previously activated with 3.0 mL of methanol and 2.0 mL of water. After sample loading, the cartridge was washed with 2.0 mL of 5% aqueous methanol. Finally, TCs were eluted with 3.0 mL of methanol. The solvent was removed under nitrogen stream and the residue was dissolved in 1.0 mL of methanol, filtered through a 0.45 μm filter, and injected into the HPLC system for its analysis.
Results and discussion
Characterization
The N2 adsorption-desorption isotherm and the correspondent BJH pore size distribution of the magnetic support are shown in Fig. 2. The material gives type IV isotherm with a sharp capillary condensation step in the medium range of the relative pressure, which is characteristic of the mesoporous materials with uniform pore size distribution. The type of hysteresis loop was H1 and it is attributed to the cylindrical pores open at both ends. The BET specific surface area, pore volume, and pore diameter were 346 m2g−1, 0.6 cm3 g−1 and 7.0 nm, respectively. The surface area in pure silica is larger than the obtained in the magnetic support, however the iron oxide incorporation decreases the porosity and the surface area [17].

a. N2 adsorption-desorption isotherm and b. pore size distribution of the magnetic support
The particle size distribution for the magnetic support is shown in Fig. 3. The powders synthesized encompass a broad range of xerogel particles ranging from 1.1 to 10.0 μm with the peak particle diameter of 2.0 μm. The synthesis of the magnetic support by emulsion polymerization plays an important role in the formation of homogenous mesostructure materials respect to the sol-gel process without surfactant [18].

Particle size distribution for the magnetic support
Optimization of TCs isolation by MSPE
Several factors affecting the proposed MSPE procedure required optimization. A Taguchi parameter design (TPD) was selected as the optimization method since it provides the necessary information with the minimal experimentation. TPD discriminates between control factors, treating them separately by means of special designed matrices (orthogonal arrays) in which the columns (corresponding to factors) and rows (corresponding to trials) are arranged in a conveniently fixed manner. These matrices indicate the combination of factor settings in each experiment and allow the simultaneous evaluation of several variables with the minimum number of trials. The results obtained were analyzed statistically to adjust each variable to its optimum.
Optimization of the system with TPD involves 5 steps, I) identifying the output variable to optimize, II) identifying and selecting factors that affect the system, III) selecting the appropriate orthogonal array and assigning adequate settings to the chosen factors, IV) analyzing the data and determining the optimum settings, and V) conducting a confirmatory experiment under the optimal conditions obtained.
In MSPE, the desired response is the maximum percentage recovery. The variables optimized in the procedure are the pH value, amount of magnetic support, dispersion time and the acetic acid concentration during the elution. The selected orthogonal array must have a number of columns equal or higher than the number of degrees of freedom of the system; thus an L9(34) array was used. The three settings selected for each factor were chosen bearing in mind preliminary analyses.
The TCs contain localized charges across all the pH values. Taking into account the milk pH value (6.5–6.9) and the pKa values [4] the TCs achieve an overall neutral state as zwitterions in this condition; therefore, the evaluated pH values were selected to assure their neutral charge. The magnetic support amount and the dispersion time had to be long enough to allow the highest analyte adsorption onto the support. With respect to the acetic acid methanolic solution, it has been shown in other pre-concentration methodologies that TCs were better eluted in their cationic form; therefore the effect of acidity on the elution process was evaluated [4]. The complexity of the milk matrix difficult the correct selection of noise factors; then in order to guarantee the robustness confirmatory experiment was done in spiked milk samples. Table 1 shows the settings for control factors used in the optimization experiments.
Table 2 shows the factorial design matrix and the recovery results obtained for each trial. All experiments were performed in triplicate in order to calculate the residual error; the total number of experiments was therefore 27 (9 experiments × 3 replicates). Measurements were performed with solutions containing 3.0 mg L−1 of each TC (TC, OT, CT); therefore the global concentration expressed as the sum of TCs was 9.0 mg L−1.
The results were analysed statistically to adjust each variable to its optimum with the least variability possible. All calculations were made using ANOVA TM v2.5 software. Table 3 shows the ANOVA results for these analyses. The values of the variance ratio (F) and the critical variance ratio (3.55 for a 95% confidence level) show that all the factors taken into account were critical (F calculated >F critical ). The factor with the greatest influence on the response was the acetic acid concentration, which accounted for 32.9% of the total variance of the results, followed by the support amount (30.7%). The contribution of the residual error was 0.02%, indicating the correct selection of experimental parameters.
Figure 4 shows the effects of the control factors on the output variable (% recovery), among which the sample volume is the most important. The combination of settings that allowed the highest output recovery was pH2-SA3-DT3-AAC2 (see Fig. 3). Taking into account the milk pH value, the magnetic support was added directly into the sample, this in turn minimizes sample manipulation. The highest levels found for support amount and dispersion time are associated with the parameters to achieve the adsorption equilibrium. The influence of the acetic acid concentration is clearly stated in the Fig. 3, as the recoveries were higher in presence of acidic methanolic solutions. An acetic acid concentration (1 × 10−3 mol L−1) was selected in order to decrease the magnetite dissolution. The mean recovery obtained in optimal conditions was 93.9 ± 1.8 (n = 5).

Effect of control factors on the mean TCs recovery obtained. pH, pH value; SA, sample amount; DT, dispersion time; AAC, acetic acid concentration
Analytical properties of the procedure
Using the FIAgram obtained under optimal conditions (Fig. 5), a standard curve for TC was constructed using spiked milk samples. Table 4 shows the regression parameters taken from this standard curve. The limits of detection were calculated according to IUPAC criteria [19], i.e., 3.29 se/b1, where se is the square root of the residual variance of the standard curve, and b1 is the slope. The intermediate precision of the procedure, expressed as the relative standard deviation (RSD) for six determinations (made on different days) using synthetic samples with analyte concentration of 0.30 mg L−1 was 1.6%. When spiked samples with an analyte concentration of 0.60 mg L−1 were used, the intermediate precision was 2.6%.

Recorder output obtained with the optimized methodology, providing a TCs calibration plot between 0.10 and 0.6 mg L−1. The peaks for a spiked sample at three levels are shown
The FIAgram was used to calculate the repeatability of the determinations; the RSD was below 5% for all standard solutions and samples. Under optimum conditions, 60 samples per hour can be analysed. Figure 5 shows the signals obtained during evaluation of the accuracy by determining the recovery of TCs (T+OT+CT) from a spiked blank milk sample at three levels (0.5, 1.0 and 2.0 FDA MRL). Overall TCs recoveries ranged from 91.0 to 97.0% with a R.S.D. below to 5.0% in all cases. These data confirmed that MSPE-FIA provided adequate sensitivity to process the samples.
To investigate the effect of successive MSPEs on the life time of the magnetic support, a 0.1 mg L-1 spike milk sample was continuously analyzed under optimal conditions. The recoveries decrease gradually with the reuse, despite after 5 determinations the analytical signal height showed good repeatability; the RSD was 4.3%.
The developed method was applied to the determination of TCs in 22 commercial milk samples. Three replicate determinations were carried out on each sample. Overall, two samples were found positive. In order to confirm the presence of a particular tetracycline, positive samples were also analyzed by MSPE-HPLC and SPE-HPLC for comparison. Results are displayed in Table 5 and Fig. 6. For each sample, the average of each analyte (determined by the methods) was compared by means of a two-way ANOVA. Calculated F value (0.24) not exceeded F tabulated value (F 2,12 = 3.89; p = 0.05), thus there are no significant differences between the results provided by both methods. The MSPE is a robust preconcentration technique that can be coupled even to FIA or HPLC.

Chromatograms obtained at the optimized conditions: a. TCs standard solution of 300 μg L−1, b. milk sample analyzed by SPE-HPLC y c. milk sample analyzed MSPE-HPLC. TC, tetracycline; OT, oxytetracycline; CT, chlortetracycline
Conclusions
In this study, a silica based magnetic support was synthesized and successfully applied for the separation of TCs antibiotics from milk samples. The TCs were isolated and the matrix interferences were eliminated using this support. The magnetic support can be reused up to 5 times. The extraction technique is a robust preconcentration technique that can be coupled successfully to FIA or HPLC. The methodology described (MSPE-FIA) is faster than classical sample preparation procedures such as SPE-HPLC, with a minimum sample handling, less solvent consumption, and is a promising screening methodology for routine milk samples analysis.
References
European Union Council Regulation No. 508/99. Off. J. Eur. Commun. L60 (1999) 16–52, http://europa.eu.int. Accessed 30 June 2010
US Code of Federal Regulations. (2003). Title 21, Part 556, Sections 150, 500, and 700. US Government Printing Office, Washington DC. (Chapter 1), http://www.fda.gov. Accessed 30 June 2010.
Hakuta T, Shinzawa H, Ozaki Y (2009) Practical method for the detection of tetracyclines in honey by HPLC and derivative UV-vis spectra. Anal Sci 25:1149–1150
Anderson CR, Rupp HS, Wu V (2005) Complexities in tetracycline analysis—chemistry, matrix extraction, cleanup, and liquid chromatography. J Chromatogr A 1075:23–32
Tzanavaras PD, Themelis DG (2007) Review of recent applications of flow injection spectrophotometry to pharmaceutical analysis. Anal Chim Acta 588:1–9
Lopez-Fernandez JM, Rios A (1995) Assessment of qualify of flow-injection methods used in food analysis—a review. Analyst 120:2400–2393
Ruiz-Capillas C, Jimenez-Colmenero F (2008) Determination of preservatives in meat products by flow injection analysis (FIA). Food Addit Contam 25:1167–1178
Ashworth RB (1985) Liquid chromatographic assay of tetracyclines in tissues of food-producing animals. J Assoc Off Anal Chem 68:1013–1018
Fritz JW, Zuo Y (2007) Simultaneous determination of tetracycline, oxytetracycline, and 4-epitetracycline in milk by high-performance liquid chromatography. Food Chem 105:1297–1301
Brandsteterova E, Kubalec P, Bovanova L, Simko P, Bednarikova A, Machackova L (1997) SPE and MSPD as pre-separation techniques for HPLC of tetracyclines in meat, milk and cheese. Z Lebensm Unters Forsch 205:311–315
Miranda JM, Rodriguez JA, Galan-Vidal CA (2009) Simultaneous determination of tetracyclines in poultry muscle by capillary zone electrophoresis. J Chromatogr A 1216:3366–3371
Caro E, Marce RM, Cormack PAG, Sherrington DC, Borrull F (2005) Synthesis and application of an oxytetracycline imprinted polymer for the solid-phase extraction of tetracycline antibiotics. Anal Chim Acta 552:81–86
Safarikova M, Safarik I (1999) Magnetic solid-phase extraction. J Magn Magn Mater 194:108–112
Tsai W, Huang T, Huang J, Hsue Y, Chuang H (2009) Dispersive solid-phase microextraction method for sample extraction in the analysis of four tetracyclines in water and milk samples by high-performance liquid chromatography with diode-array detection. J Chromatogr A 1216:2263–2269
Frenzel W, Oleksy-Frenzel J, Muller J (1992) Spectrophotmetric determination of phenolic compounds by flow-injection analysis. Anal Chim Acta 261:253–259
Aguilar-Arteaga K, Rodriguez JA, Miranda JM, Medina J, Barrado E (2010) Determination of non-steroidal anti-inflammatory drugs in wastewaters by magnetic matrix solid phase dispersion–HPLC. Talanta 80:1152–1157
Leofanti G, Padovan M, Tozzola G, Venturelli B (1998) Surface area and pore texture of catalysts. Catal Today 41:207–219
Barrado E, Rodriguez JA, Castrillejo Y (2006) Renewable stationary phase liquid magnetochromatography: determining aspartame and its hydrolysis products in diet soft drinks. Anal Bioanal Chem 385:1233–1240
Currie LA (1995) Nomenclature in evaluation of analytical methods including detection and quantification capabilities (IUPAC Recommendations 1995). Pure Appl Chem 67:1699–1723
Acknowledgments
The authors wish to thank the CONACyT (project 2006-61310), PROMEP (SIATA-Project), and Consellería de Economía e Industria proyecto INCITE09 261 380 PR for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rodriguez, J.A., Espinosa, J., Aguilar-Arteaga, K. et al. Determination of tetracyclines in milk samples by magnetic solid phase extraction flow injection analysis. Microchim Acta 171, 407–413 (2010). https://doi.org/10.1007/s00604-010-0428-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-010-0428-8