
Abstract
Background and purpose
Hydrogen sulfide (H2S) ameliorates hepatic ischemia and reperfusion injury (IRI), but the precise mechanism remains elusive. We investigated whether sodium hydrogen sulfide (NaHS), a soluble derivative of H2S, would ameliorate hepatic IRI, and if so, via what mechanism.
Methods
Mice were subjected to partial warm ischemia for 75 min followed by reperfusion. Either NaHS or saline was administered intravenously 10 min before reperfusion. The liver and serum were collected 3, 6, and 24 h after reperfusion.
Results
In the NaHS(−) group, severe IRI was apparent by the ALT leakage, tissue injury score, apoptosis, lipid peroxidation, and inflammation (higher plasma TNF-α, IL-6, IL-1β, IFN-γ, IL-23, IL-17, and CD40L), whereas IRI was significantly ameliorated in the NaHS(+) group. These effects could be explained by the augmented nuclear translocation of Nrf2, and the resulting up-regulation of HO-1 and thioredoxin-1. Phosphorylation of the PDK-1/Akt/mTOR/p70S6k axis, which is known to mediate pro-survival and anti-apoptotic signals, was significantly augmented in the NaHS(+) group, with a higher rate of PCNA-positive cells thereafter.
Conclusion
NaHS ameliorated hepatic IRI by direct and indirect anti-oxidant activities by augmenting pro-survival, anti-apoptotic, and anti-inflammatory signals via mechanisms involving Nrf-2, and by accelerating hepatic regeneration via mechanisms involving Akt-p70S6k.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Warm ischemia and reperfusion injury (IRI) is a major obstacle to the safe utilization of donation after cardiac death (DCD) grafts [1]. Warm ischemia causes mitochondrial dysfunction as well as ATP depletion and reactive oxygen species (ROS) production [2] and the nuclear translocation of NF-kappaB and AP-1 [3]. These triggers during ischemia propagate oxidative injury and expression of inflammatory cytokines, leading to eventual apoptosis and necrosis [1–3]. They also inhibit protein synthesis and cellular proliferation [4]. Among the acute responses, Akt plays a major role in survival, and anti-apoptotic and proliferative signals, including mTOR-p70s6k, the Bcl2 family, Cyclin D1, and STAT3 [5–7]. A clinically applicable method to modulate these signals is needed.
Hydrogen sulfide (H2S) can facilitate the phosphorylation of Akt and the nuclear translocation of NF-E2-related factor 2 (Nrf2) [8], leading to a reduction in IRI of the rat heart [9], kidney [10], lung [11], small intestine [12], and liver [13–16]. Although augmentation of the PI3K/Akt/p70S6k cascade in the myocardium [17] and in the small intestine [18] reduces IRI, the precise mechanism underlying this reduction is not yet fully understood. We investigated whether sodium hydrogen sulfide (NaHS), a soluble derivative of H2S [19], ameliorates hepatic IRI in mice, focusing on the signal transduction related to acute inflammation and resulting cellular survival/death and regeneration.
Materials and methods
Chemicals and reagents
All chemicals and reagents were of the highest grade commercially available, and purchased from Wako Pure Chemical Co., Ltd. (Osaka, Japan) unless otherwise stated. The antibodies used in this study were purchased from Cell Signaling Technology (Beverly, MA) unless otherwise stated.
Animals
This study was conducted with the approval of the Hokkaido University Committee for the Care and Use of Laboratory Animals. Male C57BL/6J mice, 10–12 weeks of age and weighing 25–30 g, were purchased from Sankyo Labo Service Corporation Inc. (Tokyo, Japan). The raising conditions, including chow, were as previously described [20].
Partial hepatic warm ischemia and reperfusion (I/R)
After overnight fasting, the animals were anesthetized by inhalation of isoflurane. The median and left lateral portal branches were clamped by an atraumatic aneurysm clip as previously described [21]. After closure of the abdomen, the animals were allowed to remain awake during ischemia. After 75 min, the liver was reperfused by removing the clip. The non-ischemic lobes were not resected.
Experimental protocol
The mice were divided into three groups of six animals each. Ten min before reperfusion, either NaHS (1 mg/kg; NaHS (+) group) or saline (NaHS (−) group) was administered intravenously. Sodium hydrogen sulfide (NaHS) was dissolved in saline just before administration. A sham-operated group, without vascular occlusion, was also established (sham group). Animals were killed 3, 6, and 24 h after reperfusion (R3, R6, and R24 h, respectively).
Sample collection
The ischemic lobes of the liver were collected and stored at −80 °C until use, or fixed in 10 % buffered formalin and embedded in paraffin. Plasma was also collected and stored at −80 °C until measurement.
Histological examination
Paraffin-embedded sections of the liver at R6 h were stained with hematoxylin and eosin. Histopathological grading was performed by a single pathologist in a blinded manner according to the grading described by Suzuki et al. [22] namely, sinusoidal congestion (0–4), vacuolization of hepatocyte cytoplasm (0–4), and parenchymal necrosis (0–4).
Plasma ALT activity
Plasma ALT activity at R6 h was evaluated by a Hitachi 7020 automatic analyzer (Hitachi, Tokyo, Japan).
Inflammatory cytokines and chemokines in plasma
The plasma levels of TNF-α, IL-6, IL-1β, IFN-γ, IL-17, IL-23, and CD40L at R3 h and R6 h were measured with a commercially available ELISA-based kit, Bioplex (Bio-Rad, Hercules, CA). Briefly, aliquots (20 μL) of the plasma were incubated with fluorescent-labeled antibodies and the fluorescence intensity was measured and expressed in pg/ml.
Western blot analysis
We minced and homogenized 50 mg of frozen tissue from R6 h in ice-cold lysis buffer containing Tris HCl 25 mM (pH 7.5), NaCl 150 mM, EDTA-2Na 5 mM, NaF 10 mM, sodium orthovanadate 10 mM, 1 % Nonidet P-40, and protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). The homogenate was centrifuged at 1,000×g for 10 min and the nuclear fraction was stored at −80 °C. The supernatant was then centrifuged at 15,000×g for 10 min and the resulting supernatant, being the cytosolic fraction, was stored at −80 °C. The protein concentration was measured using a BCA Protein Assay Kit (Thermo Scientific, Rockford, IL). The proteins were denatured by boiling at 95 °C for 5 min with an SDS sample buffer. Using Any-kD pre-cast gel (Bio-Rad), 40 micrograms of protein was applied to the standard SDS polyacrylamide gel electrophoresis (SDS-PAGE). Immunoblots were performed after transfer onto the PVDF membrane. Dilutions of 1:1000 were used for the primary antibodies: p-PDK-1, p-Akt, p-mTOR, p-p70s6k, cleaved caspase-3, HO-1 (Abcam, Cambridge, UK), TRX-1, β-actin, Nrf2 (Abcam), Lamin B1, and GAPDH. The dilution of the horseradish peroxidase-conjugated anti-rabbit IgG secondary antibody was 1:5000. Protein bands were detected by a chemiluminescent detector Chem Doc XRS® (Bio-Rad) using a chemiluminescence reagent, West Dura (Thermo Scientific). Protein levels were normalized by β-actin or GAPDH, and further normalized by the average value in the sham-operated group.
Apoptotic index
Frozen sections of the ischemic lobe at R24 h were stained by using a fluorescent TUNEL staining kit (Promega, Madison, WI) according to the manufacturer’s instructions. Briefly, they were fixed in 95 % ethanol and 4 % paraformaldehyde, rehydrated in PBS, digested with proteinase K, washed in PBS, incubated with equilibration buffer, and incubated with rTdT at room temperature for 1 h. The slides were mounted with Prolong Gold anti-fade reagent with DAPI (Molecular Probes Inc., Eugene, OR), and then examined with a BZ-9000 fluorescence microscope (Keyence Japan, Osaka, Japan). The apoptotic index was calculated as the number of TUNEL-positive cells divided by the total number of DAPI-positive cells. Four high power fields (HPFs) were observed per sample and the average of the four values was used.
Assessment of lipid peroxidation in the liver
The level of hepatic oxidative damage at R6 h was taken as the combined amounts of MDA and 4-HNE, the stable end products of lipid peroxidation, as determined using an LPO586 kit (Oxis International, Foster City, CA) [20]. We made 10 % (%w/v) homogenate of the liver with ice-cold Tris–HCl (20 mM) containing butylated hydroxytoluene (0.05 %). The MDA and 4-HNE contents were measured according to the manufacturer’s instructions. Data are expressed as the nmol 4-HNE equivalent per mg of wet tissue weight.
Immunohistochemistry of the liver
The frozen sections at R6 h were fixed in 95 % ethanol and 1 % formalin, rehydrated, permeabilized, and blocked in 3 % bovine serum albumin in PBS. After washing, the slide was incubated with rabbit polyclonal anti-Nrf2 antibody (1:250) (Abcam) for 1 h at room temperature, followed by Alexafluor488-conjugated goat anti-rabbit IgG secondary antibody (1:500) (Molecular Probes Inc.) for 45 min at room temperature. The slides were mounted with Prolong Gold anti-fade reagent with DAPI (Molecular Probes Inc.).
Immunohistochemistry with anti-proliferating cell nuclear antigen (PCNA) was performed according to the manufacturer’s instructions. Briefly, the paraffin-embedded sections were subjected to epitope retrieval by microwave treatment. Monoclonal mouse anti-PCNA antibody (M0879; DAKO Japan, Tokyo) was used at a dilution of 1:300. For visualization, streptavidin (LSAB 2 system HRP; DAKO Japan) and DAB substrate (DAKO Japan) were used. Nuclear counterstaining was performed using hematoxylin.
The cell-positivity rate for nuclear Nrf2 staining was counted in four random high-power fields. The resulting values were divided by the total number of DAPI-positive cells. In the case of PCNA, the number of PCNA-positive hepatocytes was counted and divided by the total number of hepatocytes under four random high-power fields.
Statistical analysis
Values are expressed as the mean ± SD. The Student’s t test or one-way ANOVA was used for evaluating statistical significance. Values of P < 0.05 were considered significant. Statistical analyses were performed using Stat View 5.0 for Windows (SAS Institute Inc., Cary, NC).
Results
Liver injury
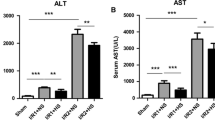
The liver histopathology appeared to be almost normal in the sham-operated group (Fig. 1a). Warm ischemia of the liver and reperfusion (hepatic warm I/R) caused inflammatory cell infiltration, congestion, and vacuolization with condensed nucleus at R6 h (Fig. 1b), whereas these changes were attenuated in the NaHS-treated mice (Fig. 1c). The injury score was augmented by hepatic warm I/R at R6 h in the NaHS(−) group, whereas it was significantly reduced by NaHS treatment (Fig. 1d). Plasma alanine aminotransferase (ALT) activity at R6 h was augmented in the hepatic warm I/R in NaHS(−) group, whereas it was significantly reduced by NaHS treatment (Fig. 1e).

Sodium hydrogen sulfide (NaHS) reduces hepatic ischemia and reperfusion injury. Mice were subjected to partial warm ischemia for 75 min and subsequent reperfusion (I/R) for 6 h. The ischemic lobe was stained with hematoxylin and eosin, and scored according to the method of Suzuki et al. Representative photographs (20× magnification) are shown. a Sham operation. b NaHS (−): I/R with vehicle treatment. c NaHS (+): I/R with NaHS (1.0 mg/kg) administration before reperfusion. d Hepatic injury score. e Plasma ALT activity 6 h after reperfusion. Results are expressed as the mean ± SD. *P < 0.05, NaHS (−) vs. NaHS (+). **P < 0.05 vs. Sham
Plasma cytokines and chemokines
Plasma levels of TNF-α (Fig. 2a), IL-6 (Fig. 2b), IL-1β (Fig. 2c), IFN-γ (Fig. 2d), IL-23 (Fig. 2e), IL-17F (Fig. 2f) and CD40L (Fig. 2g) were significantly higher in the NaHS(−) group 3 h after reperfusion (R3 h) than the respective value in the sham group, whereas the augmentation was significantly suppressed in the NaHS(+) group. By 6 h after reperfusion (R6 h), these molecules, except for CD40L, had decreased in the NaHS(−) group, and were even lower in the NaHS(+) group. Inter-group comparison revealed significant decreases in IL-6, IL-1β, IFN-γ, and IL-23, although the decreases in TNF-α and IL17F were not significant. It noteworthy that CD40L continued to rise from R3 h to R6 h in both groups, but that NaHS treatment tended to decrease its value (P = 0.065).

Sodium hydrogen sulfide (NaHS) inhibits the expression of inflammatory cytokines and chemokines. Mice were subjected to partial warm ischemia for 75 min and subsequent reperfusion (I/R). The plasma concentrations of inflammatory cytokines and chemokines at 3 and 6 h after reperfusion were measured by an ELISA-based assay. a TNF-α, b IL-6, c IL-1β, d IFN-γ, e IL-23, f IL-17F, and g soluble CD40 ligand. Results are expressed as the mean ± SD. *P < 0.05, NaHS (−) vs. NaHS (+). **P < 0.05 vs. Sham
Pro-survival signals
Pro-survival signals at R6 h were evaluated by western blots of phosphorylated PDK-1 (p-PDK1-Ser241), Akt (p-Akt-Ser473), mTOR (p-mTOR-Ser2448), and p70S6k (p-p70S6 K-Ser371). Phosphorylated PDK-1 was significantly attenuated by hepatic warm I/R, whereas the reduction was significantly less pronounced in the NaHS treatment groups (Fig. 3a). Phosphorylated Akt tended to decrease only in the NaHS(−) group, whereas in the NaHS(+) group, it was significantly higher than in the other groups (Fig. 3b). Phosphorylated mTOR and phosphorylated p70S6k were almost unchanged by hepatic warm I/R in the NaHS(−) group, whereas they were significantly higher in the NaHS treatment group than in the other groups (Fig. 3c, d).

Sodium hydrogen sulfide (NaHS) activates survival signals. Mice were subjected to partial warm ischemia for 75 min and subsequent reperfusion (I/R) for 6 h. Cytosolic protein in the ischemic lobe was applied to the western blot (top), and the relative intensity (bottom) is shown. Panels a–d show the results for a phosphorylated PDK-1 (Ser241), b phosphorylated Akt (Ser473), c phosphorylated mTOR (Ser2448), and d phosphorylated p70s6k (Ser371). Relative quantitation of each sample was performed, using GAPDH as an internal control. Each normalized value was further normalized by the mean value in the sham-operated group, and expressed as the mean ± SD. *P < 0.05, NaHS (−) vs. NaHS (+). **P < 0.05 vs. Sham
Apoptosis
TUNEL staining at R24 h showed almost no positive cells in the sham group (Fig. 4a, d). TUNEL-positive cells were augmented by hepatic warm I/R in the NaHS(−) group at R24 h (Fig. 4b, d), whereas they were significantly reduced by NaHS treatment (Fig. 4c, d). Cleaved caspase-3 at R6 h was significantly augmented by hepatic warm I/R in the NaHS(−) group, whereas it was significantly suppressed by NaHS treatment (Fig. 4e).

Sodium hydrogen sulfide (NaHS) attenuates apoptosis of hepatocytes. Mice were subjected to partial warm ischemia for 75 min and subsequent reperfusion (I/R) for 24 h. Liver sections were stained by the fluorescent TUNEL method and nuclear counterstaining with DAPI. Representative photographs (20× magnification) are shown. a Sham operation. b NaHS (−): I/R with vehicle treatment. c NaHS (+): I/R with NaHS (1.0 mg/kg) administration before reperfusion. d The apoptotic index was calculated as the number of TUNEL-positive cells divided by the number of DAPI-positive cells. Data are expressed as the mean ± SD. e Cytosolic protein of the ischemic lobe at R6 h was applied to a standard western blot. Representative western blots detected by cleaved caspase-3 antibody (top) and the ratio of the relative intensity, cleaved caspase-3/β-actin (bottom), are shown. Each normalized value was further normalized by the mean value in the sham group, and expressed as the mean ± SD. *P < 0.05, NaHS (−) vs. NaHS (+). **P < 0.05 vs. Sham
Oxidative stress
We calculated the sum of the values of malondialdehyde (MDA) and 4-hydroxy-2-nonenal (4-HNE), the end products of lipid peroxidation, at R6 h. This value was augmented by hepatic warm I/R in the NaHS(−) group, whereas it was significantly reduced by NaHS treatment (Fig. 5a).

Sodium hydrogen sulfide (NaHS) reduces oxidative stress. Mice were subjected to partial warm ischemia for 75 min and subsequent reperfusion (I/R) for 6 h. a Lipid peroxidation was assessed by MDA + 4-HNE. b Representative photographs (40× magnification) are shown. Immunohistochemistry of the liver showed that Nrf2 (Green) was ubiquitous in the cytosol but not in the nucleus in the sham-operated group. In the NaHS (−) group, only faint staining of Nrf2 in the nucleus was seen, whereas in the NaHS (+) group, almost all cells showed Nrf2-positive (pale blue; Arrow). c Western blot of nuclear proteins with regard to Nrf2. d Western blots of cytosolic proteins were evaluated for the HO-1 protein, and e thioredoxin-1 (TRX-1). Representative western blots (top) and the relative ratio (target protein/β-actin) (bottom) are shown. Each normalized value was further normalized by the mean value in the sham-operated group. Data are expressed as the mean ± SD. *P < 0.05, NaHS (−) vs. NaHS (+). **P < 0.05 vs. Sham
Anti-oxidative responses were evaluated by the translocation of Nrf2 and expression of the downstream enzymes, HO-1 and TRX-1. Immunohistochemistry of Nrf2 (Green) reveled homogeneous staining in the cytosol, but not in the nucleus, in the sham-operated group. In the NaHS (−) group, there was only faint staining of Nrf2 in the nucleus, whereas in the NaHS (+) group, almost all cells showed Nrf2-positive nucleus (pale blue; arrow), indicating that NaHS treatment augmented the nuclear translocation of Nrf2 at R6 h (Fig. 5b).
The western blot of nuclear proteins revealed that the sham group had the lowest value. The value in the NaHS(−) group at R6 h was increased significantly, and it was further increased significantly in the NaHS(+) group (Fig. 5c). The expression of HO-1 in the cytosol was increased by hepatic warm I/R in the NaHS(−) group, and it was further augmented significantly by NaHS treatment (Fig. 5d). The expression of TRX-1 in the cytosol showed a tendency to decrease with hepatic warm I/R in the NaHS(−) group, whereas it was significantly augmented by NaHS treatment (Fig. 5e).
Proliferation
Hepatic proliferation was evaluated by PCNA staining at R24 h (Fig. 6a–d). In the sham group, the percentage of PCNA-positive hepatocytes was 47 ± 14 %. The positive rate was reduced significantly to 13 ± 15 % in the NaHS(−) group, whereas it was augmented significantly to 63 ± 14 % by NaHS treatment.

Sodium hydrogen sulfide (NaHS) stimulates liver regeneration. Mice were subjected to partial warm ischemia for 75 min and subsequent reperfusion (I/R) for 24 h. Liver sections were stained by the anti-proliferating cell nuclear antigen (PCNA) antibody and hematoxylin as nuclear counterstaining. Representative photographs (20× magnification) are shown. In the sham group (a), the positive rate was 47 %, but it was significantly decreased in the NaHS(−) group (b), and it was significantly augmented in the NaHS(+) group (c). The rate of PCNA-positive hepatocytes is shown in (d). Values are expressed as the mean ± SD. *P < 0.05, NaHS (−) vs. NaHS (+). **P < 0.05 vs. Sham
Discussion
We confirmed the beneficial effects of NaHS against warm I/R of the mouse liver, by demonstrating a reduction in tissue injury, apoptosis, oxidative damage, and inflammatory reactions, with stimulation of liver regeneration. These beneficial effects were at least in part due to the augmented nuclear translocation of Nrf2 and downstream activation of anti-inflammatory and anti-oxidant pathways. Activation of the pro-survival signals was demonstrated by the augmented phosphorylation of PDK-1, Akt, mTOR, and p70s6k in response to NaHS treatment. Simultaneous activation of anti-apoptotic, anti-inflammatory, anti-oxidative, pro-survival, and pro-proliferation cascades appeared to allow recovery of the liver subjected to warm I/R.
Hydrogen sulfide (H2S) is produced endogenously from cysteine in the liver, kidney, vessels, brain, and nerves, and its exertion at low concentrations is biologically important [23]. Furthermore, H2S has been reported to reduce IRI of the liver [13–16] and other organs [9–12]. Consistent with these reports, the present study showed a reduction in net injury by ALT leakage and histopathology.
Acute inflammation in hepatic IRI is initiated mainly in Kupffer cells and hepatocytes during warm ischemia [2, 3, 24]. These cells release TNF-α, IL-6, and IL-1β, and stimulate inflammation, which in turn activates the cell death pathway, and ROS and protease release from neutrophils [3, 4]. Schlegel et al. [25] reported that serum TNF-α, IL-17, and the ratio of CD154-positive T cells were increased in a DCD liver graft after transplantation. IL-23 and Th17 cells, including NK, NKT, and γδT cells, play major roles in both acquired and innate immunity in organ transplantation [26]. In hepatic IRI, IL-23 stimulates Kupffer cells and CD4 +/Th17 cells. IL-6 released from Kupffer cells promotes further activation of Th17 cells to release IL-17, and stimulates neutrophil accumulation [27]. A recent report revealed that activation of CD40-CD40L (CD154) promoted oxidative stress-induced apoptosis in hepatocytes [28].
In this study, serum TNF-α, IL-6, IL-1β, IFN-γ, IL-17, IL-23, and soluble CD40L (CD154) increased significantly within 3 h of reperfusion, but these changes were inhibited by NaHS. Hydrogen sulfide reduced hepatic IRI with less production of TNFα and IL-6 [16]. Suppression of the IL23–IL17 axis reduced hepatic IRI [29]. Altogether, these facts suggest that NaHS reduced hepatic IRI by inhibiting the IL23–IL17 axis, thereby inhibiting the activation of Kupffer cells, neutrophils, and lymphocytes (CD4 +/Th17 cells) in the early phase of reperfusion. Although there are various sources of CD40L, the main source is the activated platelets [30], and CD40L was found to worsen hepatic IRI [28]. In this study, there was a massive release of cytokines and chemokines at R3 h. Multiple pro-inflammatory mediators from various cell types, platelet-endothelial adhesion, and platelet-leukocyte aggregation would stimulate the further activation of platelets [2], leading to the sustained high level of soluble CD40L.
Oxidative stress is another important factor in hepatic IRI [2–4]. Oxidative damage in the sinusoidal endothelial cells stimulates endothelin-1 (ET-1) production, leading to microcirculatory disturbance [31, 32]. Several distinct mechanisms of the anti-oxidant property of H2S have been reported, namely, direct scavenging of ROS [33], reversible inhibition of mitochondrial respiration [34], augmentation of glutathione (GSH) production through enhanced cystine/cysteine transport [35], and Nrf2-dependent expression of anti-oxidant and anti-inflammatory proteins [23]. Consistent with these reports, NaHS reduced lipid peroxidation and inflammation in this study.
Nrf2 exists in the cytosol with Keap-1 in the resting state, but it dissociates from Keap-1 on exposure to stimuli, such as oxidative stress and pro-survival signals, leading to up-regulation of HO-1 [36] and TRX-1 [37]. HO-1 reduces oxidative stress through the conversion of heme into biliverdin and carbon monoxide (CO) [38]. Furthermore, CO exerts anti-inflammatory and vasodilatory effects [39] and TRX-1 reduces protein thiol and/or hydrogen peroxide with the support of GSH [40]. In line with these reports, our study showed, for what we believe to be the first time, that NaHS augmented HO-1 and TRX-1 levels through augmented nuclear translocation of Nrf2, leading to reductions in oxidative stress and inflammation.
The anti-apoptotic and pro-survival effects of H2S against mitochondria would result in an inhibition of intrinsic apoptosis [36]. H2S activates this pathway in the I/R of hippocampal neurons [7, 41] and myocardium [5]. Insulin supplementation has been shown to stimulate myocardial surviving expression via activation of PI3K-Akt-mTOR-p70s6k, resulting in anti-apoptotic effects [42]. In this study, pro-survival signals mediated by the PDK-1-Akt-mTOR-p70s6k axis were maintained by the higher phosphorylation in the NaHS-treated liver. Phosphorylation of p70s6k confers protection against IRI in the heart and small intestines through anti-apoptotic and anti-inflammatory effects [17, 18]. The enhanced PDK1-Akt signal reduced IRI through the augmented phosphorylation of PDK1 [43] and Akt [14], but not through de novo gene expression of these proteins, at least within 6 h of reperfusion. In line with these reports, this study is the first to show that NaHS supplementation ameliorated hepatic warm IRI by maintaining the phosphorylation of p70s6k and upstream kinases, including mTOR, Akt, and PDK-1. Since we did not assess any gene expression, further study is required to clarify the precise mechanism of NaHS-mediated protection during the early phase of reperfusion.
Another important anti-apoptotic signal in hepatic IRI is the signal transducer and activator of transcription 3 (STAT3) [9]. Ke et al. [29] recently reported that HO-1 ameliorated hepatic inflammation, apoptosis, and net injury after warm I/R by inhibiting NF-kappaB signals in the nucleus. They showed that STAT3 was indispensable for the HO-1-mediated down-regulation of TLR-4 and PTEN, and the augmentation of phospho-Akt. In the present study we observed a transient rise of IL-6 at 3 h, only slight increases of IL-6 and TNF-α at 6 h, and a significantly higher PCNA-positivity rate 24 h after reperfusion in the NaHS-treated group. Debonera et al. [44] reported that IL-6-mediated activation of STAT3 did not trigger liver regeneration in severely injured liver grafts. Although we did not evaluate STAT3 here, these observations suggest that the STAT3-mediated machinery of liver regeneration [45] would have functioned well in the NaHS-treated liver.
Recently, Zhang et al. [46] reported that NaHS administration before ischemia inhibited mitochondrial permeability transition pore opening and activated Akt-GSK3β. Exogenously administered hydrogen sulfide disappeared rapidly from the blood and tissues through oxidation and thiol-binding [47], with half-lives of 2.0 and 5.4 min in the aerobic and anaerobic liver, respectively [48]. Therefore, we administered NaHS before reperfusion to maintain enough concentration at reperfusion. In contrast to the previous report [46], we failed to show the efficacy by administration before ischemia in our preliminary study. In relation to dosage, Kang et al. [15] reported the effective dose to be 0.78 mg/kg. Since 0.5 mg/kg was less effective than 1 mg/kg, and 3 mg/kg resulted in animal death by respiratory dysfunction in our preliminary study, we adopted 1 mg/kg as the optimal dose (data not shown). The controversy might be due to the differences in ischemia time, species, and strain. Although further investigation is necessary to establish the optimal mode of administration for liver graft protection, it is encouraging that hydrogen sulfide proved effective when administered before ischemia [46], before reperfusion, and during cold preservation [49].
In conclusion, NaHS treatment against hepatic warm I/R resulted in high phosphorylation levels of PDK1, Akt, mTOR, and p70S6k, and nuclear translocation of Nrf2, leading to anti-oxidant, anti-inflammatory, anti-apoptotic, pro-survival, and pro-proliferative effects, and eventually reduced net IRI with rapid liver regeneration.
Abbreviations
- ALT:
-
Alanine aminotransferase
- CO:
-
Carbon monoxide
- DCD:
-
Donation after cardiac death
- ELISA:
-
Enzyme-linked immunosorbent assay
- ER:
-
Endoplasmic reticulum
- GSH:
-
Glutathione
- 4-HNE:
-
4-hydroxy-2-nonenal
- HO-1:
-
Heme oxygenase 1
- TRX-1:
-
Thioredoxin-1
- HPFs:
-
High-power fields
- H2S:
-
Hydrogen sulfide
- HSPs:
-
Heat shock proteins
- IL-6:
-
Interleukin 6
- I/R:
-
Ischemia and reperfusion
- IRI:
-
Ischemia and reperfusion injury
- Keap-1:
-
Kelch-like ECH-associated protein 1
- MDA:
-
Malondialdehyde
- MPT:
-
Mitochondrial permeability transition
- mTOR:
-
Mammalian target of rapamycin
- NaHS:
-
Sodium hydrogen sulfide
- NF-kappaB:
-
Nuclear factor-kappa B
- Nrf2:
-
NF-E2-related factor 2
- PDK-1:
-
Phosphoinositide-dependent kinase 1
- PI3K:
-
Phosphoinositide 3 kinase
- PKC:
-
Protein kinase C
- PNF:
-
Primary graft non-function
- PVDF:
-
Polyvinylidene difluoride
- p70s6k:
-
70-kDa Ribosomal protein S6 kinase
- ROS:
-
Reactive oxygen species
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- STAT3:
-
Signal transducer and activator of transcription 3
- TNF-α:
-
Tumor necrosis factor α
- TRX-1:
-
Thioredoxin 1
- TUNEL:
-
Terminal dUTP nick end-labeling
References
Monbaliu D, Pirenne J, Talbot D. Liver transplantation using donation after cardiac death donors. J Hepatol. 2012;56:474–85.
Vollmar B, Menger MD. The hepatic microcirculation: mechanistic contributions and therapeutic targets in liver injury and repair. Physiol Rev. 2009;89:1269–339.
Zwacka RM, Zhou W, Zhang Y, Darby CJ, Dudus L, Halldorson J, et al. Redox gene therapy for ischemia/reperfusion injury of the liver reduces AP1 and NF-kappaB activation. Nat Med. 1998;4:698–704.
Abu-Amara M, Yang SY, Tapuria N, Fuller B, Davidson B, Seifalian A. Liver ischemia/reperfusion injury: processes in inflammatory networks—a review. Liver Transpl. 2010;16:1016–32.
Hu Y, Chen X, Pan TT, Neo KL, Lee SW, Khin ES, et al. Cardioprotection induced by hydrogen sulfide preconditioning involves activation of ERK and PI3 K/Akt pathways. Pflugers Arch. 2008;455:607–16.
Tsang A, Hausenloy DJ, Mocanu MM, Yellon DM. Postconditioning: a form of “modified reperfusion” protects the myocardium by activating the phosphatidylinositol 3-kinase-Akt pathway. Circ Res. 2004;95:230–2.
Noshita N, Lewen A, Sugawara T, Chan PH. Evidence of phosphorylation of Akt and neuronal survival after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2001;21:1442–50.
King AL, Lefer DJ. Cytoprotective actions of hydrogen sulfide in ischaemia-reperfusion injury. Exp Physiol. 2011;96(9):840–6.
Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, et al. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ Res. 2009;105:365–74.
Hunter JP, Hosgood SA, Patel M, Rose R, Read K, Nicholson ML. Effects of hydrogen sulphide in an experimental model of renal ischaemia-reperfusion injury. Br J Surg. 2012;99:1665–71.
Fu Z, Liu X, Geng B, Fang L, Tang C. Hydrogen sulfide protects rat lung from ischemia-reperfusion injury. Life Sci. 2008;82:1196–202.
Henderson PW, Weinstein AL, Sohn AM, Jimenez N, Krijgh DD, Spector JA. Hydrogen sulfide attenuates intestinal ischemia-reperfusion injury when delivered in the post-ischemic period. J Gastroenterol Hepatol. 2010;25:1642–7.
Wang D, Ma Y, Li Z, Kang K, Sun X, Pan S, et al. The role of AKT1 and autophagy in the protective effect of hydrogen sulphide against hepatic ischemia/reperfusion injury in mice. Autophagy. 2012;8:954–62.
Jha S, Calvert JW, Duranski MR, Ramachandran A, Lefer DJ. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: role of antioxidant and antiapoptotic signaling. Am J Physiol Heart Circ Physiol. 2008;295:H801–6.
Kang K, Zhao M, Jiang H, Tan G, Pan S, Sun X. Role of hydrogen sulfide in hepatic ischemia-reperfusion-induced injury in rats. Liver Transpl. 2009;15:1306–14.
Bos EM, Snijder PM, Jekel H, Weij M, Leemans JC, van Dijk MC, et al. Beneficial effects of gaseous hydrogen sulfide in hepatic ischemia/reperfusion injury. Transpl Int. 2012;25:897–908.
Kis A, Yellon DM, Baxter GF. Second window of protection following myocardial preconditioning: an essential role for PI3 kinase and p70S6 kinase. J Mol Cell Cardiol. 2003;35:1063–71.
Ban K, Kozar RA. Protective role of p70S6 K in intestinal ischemia/reperfusion injury in mice. PLoS One. 2012;7:e41584.
Lowicka E, Beltowski J. Hydrogen sulfide (H2S)—the third gas of interest for pharmacologists. Pharmacological reports. 2007;59:4–24.
Fukai M, Hayashi T, Yokota R, Shimamura T, Suzuki T, Taniguchi M, et al. Lipid peroxidation during ischemia depends on ischemia time in warm ischemia and reperfusion of rat liver. Free Radic Biol Med. 2005;38:1372–81.
Yadav SS, Gao W, Harland RC, Clavien PA. A new and simple technique of total hepatic ischemia in the mouse. Transplantation. 1998;65:1433–6.
Suzuki S, Nakamura S, Koizumi T, Sakaguchi S, Baba S, Muro H, et al. The beneficial effect of a prostaglandin I2 analog on ischemic rat liver. Transplantation. 1991;52:979–83.
Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–35.
Nakamitsu A, Hiyama E, Imamura Y, Matsuura Y, Yokoyama T. Kupffer cell function in ischemic and nonischemic livers after hepatic partial ischemia/reperfusion. Surg Today. 2001;31:140–8.
Schlegel A, Graf R, Clavien PA, Dutkowski P. Hypothermic oxygenated perfusion (HOPE) protects from biliary injury in a rodent model of DCD liver transplantation. J Hepatol. 2013;59:984–91.
Chen Y, Wood KJ. Interleukin-23 and TH17 cells in transplantation immunity: does 23 + 17 equal rejection? Transplantation. 2007;84:1071–4.
Husted TL, Blanchard J, Schuster R, Shen H, Lentsch AB. Potential role for IL-23 in hepatic ischemia/reperfusion injury. Inflamm Res. 2006;55:177–8.
Bhogal RH, Weston CJ, Curbishley SM, Adams DH, Afford SC. Activation of CD40 with platelet derived CD154 promotes reactive oxygen species dependent death of human hepatocytes during hypoxia and reoxygenation. PLoS One. 2012;7:e30867.
Ke B, Shen XD, Ji H, Kamo N, Gao F, Freitas MC, et al. HO-1-STAT3 axis in mouse liver ischemia/reperfusion injury: regulation of TLR4 innate responses through PI3 K/PTEN signaling. J Hepatol. 2011;56:359–66.
Shen X, Wang Y, Gao F, Ren F, Busuttil RW, Kupiec-Weglinski JW, et al. CD4 T cells promote tissue inflammation via CD40 signaling without de novo activation in a murine model of liver ischemia/reperfusion injury. Hepatology. 2009;50:1537–46.
Yokota R, Fukai M, Shimamura T, Suzuki T, Watanabe Y, Nagashima K, et al. A novel hydroxyl radical scavenger, nicaraven, protects the liver from warm ischemia and reperfusion injury. Surgery. 2000;127:661–9.
Ota T, Hirai R, Urakami A, Soga H, Nawa S, Shimizu N. Endothelin-1 levels in portal venous blood in relation to hepatic tissue microcirculation disturbance and hepatic cell injury after ischemia/reperfusion. Surg Today. 1997;27:313–20.
Johansen D, Ytrehus K, Baxter GF. Exogenous hydrogen sulfide (H2S) protects against regional myocardial ischemia-reperfusion injury–Evidence for a role of K ATP channels. Basic Res Cardiol. 2006;101:53–60.
Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci USA. 2007;104:15560–5.
Kimura Y, Goto Y, Kimura H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid Redox Signal. 2010;12:1–13.
Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem Pharmacol. 2010;80:1895–903.
Kim YC, Yamaguchi Y, Kondo N, Masutani H, Yodoi J. Thioredoxin-dependent redox regulation of the antioxidant responsive element (ARE) in electrophile response. Oncogene. 2003;22:1860–5.
Akamatsu Y, Haga M, Tyagi S, Yamashita K, Graça-Souza AV, Ollinger R, et al. Heme oxygenase-1-derived carbon monoxide protects hearts from transplant associated ischemia reperfusion injury. Faseb J. 2004;18:771–2.
Wei Y, Chen P, de Bruyn M, Zhang W, Bremer E, Helfrich W. Carbon monoxide-releasing molecule-2 (CORM-2) attenuates acute hepatic ischemia reperfusion injury in rats. BMC Gastroenterol. 2010;10:42.
Watanabe R, Nakamura H, Masutani H, Yodoi J. Anti-oxidative, anti-cancer and anti-inflammatory actions by thioredoxin 1 and thioredoxin-binding protein-2. Pharmacol Ther. 2010;127:261–70.
Shao JL, Wan XH, Chen Y, Bi C, Chen HM, Zhong Y, et al. H2S protects hippocampal neurons from anoxia-reoxygenation through cAMP-mediated PI3 K/Akt/p70S6 K cell-survival signaling pathways. J Mol Neurosci. 2011;43:453–60.
Si R, Tao L, Zhang HF, Yu QJ, Zhang R, Lv AL, et al. Survivin: a novel player in insulin cardioprotection against myocardial ischemia/reperfusion injury. J Mol Cell Cardiol. 2011;50:16–24.
Koh PO. Melatonin prevents hepatic injury-induced decrease in Akt downstream targets phosphorylations. J Pineal Res. 2011;51:214–9.
Debonera F, Wang G, Xie J, Que X, Gelman A, Leclair C, et al. Severe preservation injury induces Il-6/STAT3 activation with lack of cell cycle progression after partial liver graft transplantation. Am J Transplant. 2004;4:1964–71.
Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol. 2004;5:836–47.
Zhang Q, Fu H, Zhang H, Xu F, Zou Z, Liu M, et al. Hydrogen sulfide preconditioning protects rat liver against ischemia/reperfusion injury by activating Akt-GSK-3β signaling and inhibiting mitochondrial permeability transition. PLoS One. 2013;8:e74422.
Klingerman CM, Trushin N, Prokopczyk B, Haouzi P. H2S concentrations in the arterial blood during H2S administration in relation to its toxicity and effects on breathing. Am J Physiol Regul Integr Comp Physiol. 2013;305:R630–8.
Vitvitsky V, Kabil O, Banerjee R. High turnover rates for hydrogen sulfide allow for rapid regulation of its tissue concentrations. Antioxid Redox Signal. 2012;17:22–31.
Balaban CL, Rodriguez JV, Guibert EE. Delivery of the bioactive gas hydrogen sulfide during cold preservation of rat liver: effects on hepatic function in an ex vivo model. Artif Organs. 2011;35:508–15.
Acknowledgments
We thank Mr. Masatoshi Horigome and Ms. Sayaka Miyoshi for their excellent technical support. This work was supported in part by a Public Trust Surgery Fund (2012) and a grant-in aid for Scientific Research from the Ministry of Education, Science, Sports, and Culture of Japan (No. 25293272).
Conflict of interest
Shingo Shimada and his co-authors have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Shimada, S., Fukai, M., Wakayama, K. et al. Hydrogen sulfide augments survival signals in warm ischemia and reperfusion of the mouse liver. Surg Today 45, 892–903 (2015). https://doi.org/10.1007/s00595-014-1064-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00595-014-1064-4