
Abstract
Epigenetic alterations, such as DNA methylation, histone modification and the loss of genome imprinting, are important indicators of human carcinogenesis. DNA methylation is a fundamental epigenetic process that modulates the gene expression levels. In cancer cells, DNA methylation may be altered in two principle ways: global DNA hypomethylation and site-specific CpG island promoter hypermethylation. Long interspersed element-1 (LINE-1 or L1) is a repetitive DNA retrotransposon that duplicates via a copy-and-paste genetic mechanism. Since LINE-1 constitutes a substantial portion (approximately 17 %) of the human genome, the extent of LINE-1 methylation is regarded to be a surrogate marker of global DNA methylation. Measuring the level of LINE-1 methylation using pyrosequencing technology has emerged as a cost-effective and high-throughput method for assessing the global DNA methylation status. In some types of gastrointestinal (GI) cancers, LINE-1 hypomethylation is strongly associated with a poor prognosis, supporting its potential role as a prognostic marker. In addition, the LINE-1 methylation level may prove to be a useful clinical biomarker for assessing the risk of cancer or predicting the chemotherapeutic efficacy of treatment in patients with GI cancers. In this article, we summarize current knowledge regarding LINE-1 methylation and its clinical implications in GI cancers, including colorectal cancer, gastric cancer and esophageal cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
One of the most widely studied epigenetic alterations is DNA methylation, the covalent postreplicative addition of a methyl group (–CH3) to the 5-carbon of the cytosine ring in CpG dinucleotides. CpG dinucleotides are non-uniformly distributed throughout the human genome [1–3] and typically occur at a frequency of approximately one per 80 dinucleotides. Meanwhile, isolated CpG-rich regions [with a length of 200 base pairs (bps) to several kilobases (kbs)], referred to as CpG islands, comprise 1–2 % of the human genome. An estimated 45,000 CpG islands lie close to the promoter regions of various genes. DNA hypermethylation at promoter CpG sites can silence the expression of tumor suppressor genes, thus contributing to the development and progression of cancer [3].
The second important DNA methylation alteration in human neoplasms is genome-wide DNA hypomethylation [4]. The causal mechanisms of this process remain unknown. However, global DNA hypomethylation may contribute to multistep tumorigenesis in several ways. Experimental studies indicate that DNA hypomethylation of repetitive sequences [i.e. short interspersed transposable elements (SINE or Alu elements) or long interspersed transposable elements (LINEs)] may predispose cells to chromosomal defects and rearrangements, resulting in genetic instability [5]. Such increases in chromosomal instability result in the development and progression of cancer. Since LINE-1 constitutes a substantial portion (approximately 17 %) of the human genome [6], the level of LINE-1 methylation is regarded to be a surrogate marker of global DNA methylation. Importantly, LINE-1 hypomethylation has emerged as a promising prognostic or predictive biomarker in several types of human neoplasms [7, 8].
In this review, we summarize the accumulated evidence supporting the use of the LINE-1 methylation level as an indicator of gastrointestinal cancers (i.e. colorectal, gastric and esophageal cancer).
Long interspersed transposable element-1 (LINE-1)
Repetitive sequences account for half of the human genome and are subdivided into two principal types [9, 10]. The first is the tandem repeat, or satellite, in which each repeat unit is immediately adjacent to other units [11]. The second consists of interspersed repeats, repeated sequences that are distributed throughout the genome [12]. Interspersed repeats are derived from transposable elements or mobile DNAs. The transposable elements comprise DNA transposons and retrotransposons. The former elements move via a “cut-and-paste mechanism” mediated by an element-encoded transposase. Retrotransposons are DNA sequences that transpose through an RNA intermediate via a “copy-and-paste mechanism.” They are subdivided into sequences containing long terminal repeats (LTR) and sequences that lack these features (non-LTR). Most of the mobile element activity in humans appears to involve non-LTR retrotransposons, typified by long interspersed element-1 (LINE-1 or L1). LINE-1 has been amplified in the human genome to more than 500,000 copies, or 17 % of the genomic sequence [9]. LINE-1 consists of two open reading frames (ORF1 and ORF2) flanked by a 5′- and 3′-untranslated region (UTR). ORF1 encodes an RNA-binding protein, while ORF2 encodes a protein with endonuclease and reverse-transcriptase activities [13]. LINE-1 is present on both homologous chromosomes and is truncated (mean length = 0.9 kb) in the human genome. A limited portion of LINE-1 is present in a potentially active form with full-length elements (~6 kb). Therefore, LINE-1 is often regarded to be a non-informative miscellany of the genetic past and has been referred to as “parasitic” or “junk” DNA. However, accumulating evidence suggests a crucial role of LINE-1 in various cellular processes. LINE-1 can continuously rearrange the genome, thereby influencing the gene expression status in different ways. LINE-1 may induce genetic variation and polymorphism via recombination and rearrangement as well as in-house mutagenesis [14–16]. In addition, the LINE-1 expression may contribute to transcription disruptions, insertion mutations or DNA breaks, leading to genomic instability in cancer cells [17]. The antisense promoter of LINE-1 may also change the gene expression status by influencing the transcriptional activity of surrounding genes [18–20].
Methylation of LINE-1 repetitive elements
Reportedly, more than one-third of DNA methylation occurs in repetitive sequences. Full-length LINE-1 has an internal promoter in its 5′UTR, which ranges from +1 to 909 bp [21]. The initial 460-bp region of the 5′UTR includes 29 CpG sites, whose methylation status has been extensively investigated and shown to be high [22–24]. Interestingly, the LINE-1 methylation levels in normal tissues strongly depend on the tissue type; the range of the LINE-1 methylation levels is narrow in some tissues (such as the liver, kidneys, breast, stomach and lungs) and wide in other tissues (such as the esophagus and thyroid) [25]. The LINE-1 methylation status has also been proposed to vary at individual genomic locations. One study of human cancer cell lines demonstrated that the methylation levels differ at nine LINE-1 loci [26]. Another study that evaluated the methylation patterns of 17 LINE-1 loci in several cell types found that the methylation levels at these loci are influenced differentially, depending on the location of the particular sequences in the genome [27]. Collectively, the changing methylation status observed in different sets of LINE-1 loci may lead to different cellular phenotypes.
Assays for evaluating the LINE-1 methylation levels
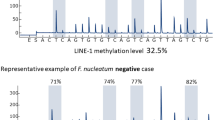
CpG methylation analyses basically discriminate between methylated and unmethylated DNA sequences, usually following PCR amplification of the targeted sequence [28]. Commonly used post-PCR detection techniques for performing LINE-1 assays include the Southern blot, combined bisulfite restriction analysis (COBRA), methylation-specific PCR after bisulfite treatment (MSP) and pyrosequencing [29–32]. Pyrosequencing is a non-electrophoretic nucleotide extension sequencing technology designed for various applications, including single-nucleotide polymorphism genotyping, bacterial strain typing, mutation detection in tumors and quantitative CpG island methylation analyses. Pyrosequencing is a promising clinical tool because it is accurate, reliable, easily interpreted and feasible for use in LINE-1 methylation detection. Aparicio et al. [33] demonstrated higher reproducibility and lower variability of pyrosequencing relative to COBRA, methylation-sensitive single-nucleotide-primer extension (MsSNuPE) and Methylight. Irahara et al. [34] showed that LINE-1 pyrosequencing assays are precise and can be used to reliably measure LINE-1 methylation in paraffin-embedded colon cancer tissues, normal colon tissues and peripheral blood cells. Our group also confirmed the reliability and reproducibility of LINE-1 assays of paraffin-embedded esophageal cancer tissues and cancer cell lines (Fig. 1). We performed bisulfite conversion on five different DNA specimen aliquots (bisulfite-to-bisulfite) and conducted five sequential runs of PCR pyrosequencing. The bisulfite-to-bisulfite standard deviation (SD) ranged from 1.44 to 2.90, while the SD of a series of runs ranged from 0.63 to 3.25 [35]. These studies suggest that bisulfite conversion and PCR-pyrosequencing assays can be used to accurately measure LINE-1 methylation in human cancers and may be useful in clinical and research settings. Indeed, these assays have been applied in large study groups by independent research teams [36–39].

Pyrosequencing assay for measuring the LINE-1 methylation levels. Pyrograms of two GI cancer cell lines (HCT116 and MKN1) are shown. The percentages indicate the proportion of cytosines at each CpG site following bisulfite conversion that was used to estimate the methylation level at each CpG site. Overall, the LINE-1 methylation level is the average of the proportions of cytosines (%) at the four CpG sites. The X axis shows the dispensation order. The height of each peak (Y axis) is proportional to the number of nucleotides incorporated. Nucleotide incorporation generates light observed as a peak in the pyrogram trace
LINE-1 methylation levels in various cancers
In 1983, Feinberg et al. [40] first reported that the genes of cancer cells are substantially more hypomethylated than their normal counterparts. The methylation status of LINE-1 in cancer cell lines was first demonstrated by Thayer et al. [22] in 1993. Since then, tumoral LINE-1 hypomethylation has been reported in many types of human tumors, including those of the prostate [41–43], ovaries [44, 45], head and neck [39, 46, 47], lungs [48, 49], urothelium [50, 51], blood (leukemia [52, 53]), oral cavity [54], thyroid [55], breast [56] and testicles [57]. Using a wide range of cancer cell lines and clinical specimens, researchers have demonstrated wide variability in the LINE-1 hypomethylation levels among normal and cancer tissue types [25, 27, 30, 58, 59]. However, some studies of thyroid cancer [55], renal tumors [60], lymphoma and leukemia [61] reported no changes in the LINE-1 methylation levels. In certain types of human neoplasms, LINE-1 hypomethylation is associated with a poor clinical outcome (Table 1). In contrast, a relationship between LINE-1 hypomethylation and a favorable prognosis has been demonstrated in patients with melanoma [62]. This discrepancy may be due to differences in the tumor histological type. The mechanism by which LINE-1 hypomethylation affects aggressive tumor behavior has yet to be revealed. One possible mechanism involves the link between LINE-1 hypomethylation and genomic instability. Gaudet et al. generated transgenic mice carrying a hypomorphic DNA methyltransferase 1 (Dnmt1). The mutant mice displayed substantial genome-wide hypomethylation in all tissues. They also developed aggressive T cell lymphomas, with a high frequency of chromosome 15 trisomy; thus suggesting that DNA hypomethylation plays a causal role in tumor formation by promoting chromosomal instability [5]. Igarashi et al. [63] demonstrated a significant correlation between LINE-1 hypomethylation and chromosomal aberrations in gastrointestinal stromal tumors using array-based comparative genomic hybridization (array CGH). We are currently performing further studies utilizing array CGH to clarify the relationships between LINE-1 hypomethylation, genomic aberrations and patient survival in esophageal cancer. Another possible mechanism is transcriptional dysregulation, which possibly activates proto-oncogenes, endogenous retroviruses and transposable elements that affect tumor aggressiveness. Given the epigenetic regulation of microRNA in human cancers [64], global DNA hypomethylation may contribute to the acquisition of aggressive tumor behavior via an aberrant microRNA expression. Furthermore, in addition to acting as a surrogate marker of global DNA methylation, the LINE-1 methylation status itself likely exerts biological effects. Recall that LINE-1 elements are retrotransposons, which provide alternative promoters and contribute to the non-coding RNA expression, thereby regulating the functions of multiple genes. Further studies are therefore needed to elucidate the mechanism(s) by which LINE-1 hypomethylation affects tumor behavior.
LINE-1 methylation in colorectal cancer
Most research on the LINE-1 methylation levels in GI cancers has focused on colorectal cancer (Table 2), in which the levels of LINE-1 methylation are variably reduced. In Ogino et al.’s [65] study, the LINE-1 methylation levels were found to be widely and approximately normally distributed [mean 61.4 (%), median 62.3, SD 9.6, range 23.1–90.3] in a cohort of 869 colorectal cancer patients. The levels of LINE-1 methylation in colorectal cancer cell lines (COLO205, HCC-2998, HCT116, HCT15, HT29, KM12, LOVO, RKO, SW48, SW620) are also highly variable, ranging from 30 to 70 % [58].
The microsatellite instability (MSI) pathway is recognized to be a pathway of colorectal cancer development [66]. LINE-1 hypomethylation is inversely related to the MSI status [58, 65]. Goel et al. [67] demonstrated that the level of LINE-1 methylation in microsatellite stable, Amsterdam-positive hereditary nonpolyposis colorectal cancer (MSS HNPCC) tumors is significantly lower than that observed in other types of cancers (Lynch syndrome cancers, sporadic MSS cancers, sporadic MSI cancers). In colorectal cancer, several tumor suppressor genes are silenced by promoter CpG island methylation [68]. A subset of colorectal cancers exhibit widespread promoter CpG island methylation, referred to as the CpG island methylator phenotype (CIMP) [68–70]. An inverse relationship between LINE-1 hypomethylation and CIMP has also been reported [65]. These findings suggest that CIMP/MSI and genomic hypomethylation represent different pathways to the development of colorectal cancer.
The relationship between LINE-1 hypomethylation and early-onset colorectal cancer is intriguing. Early-onset colorectal cancer presents a clinically distinct colorectal cancer phenotype and is often associated with an unfavorable prognosis. LINE-1 extreme hypomethylators (methylation < 40 %) appear significantly more frequently in younger than in older patients [71]. Antelo et al. [72] identified a similar relationship between early-onset colorectal cancer (≤50 years of age) and LINE-1 hypomethylation. Collectively, LINE-1 hypomethylation may be a potentially important indicator of early-onset colorectal cancer.
Whether the LINE-1 methylation level in colorectal cancers is associated with tumor stage progression remains controversial. Sunami et al. [73] showed that LINE-1 demethylation progression is linearly correlated with that of tumor node metastasis (TNM). However, Ogino et al. [65], in a study of 869 population-based colorectal cancer tumors, reported no relationships between the LINE-1 methylation levels and tumor stage (stage I–IV). Matsunoki et al. [74] reported that the LINE-1 methylation levels are identical between primary tumors and liver/LN metastases and that the LINE-1 methylation levels in primary tumors are homogeneous, which is in agreement with our recent findings [75]. In addition, our group reported that the LINE-1 methylation levels do not differ between synchronous tumors and metachronous tumors, supporting the hypothesis that the LINE-1 methylation level is preserved throughout the long-term natural history of colorectal cancer development [75]. The results of Ogino et al., Matsunoki et al. and our group suggest that LINE-1 hypomethylation is initiated at an early stage of cancer development and that the LINE-1 methylation level remains relatively stable during tumor progression. In contrast, Hur et al. showed that the LINE-1 methylation levels in liver metastases from colorectal cancer are significantly lower than those observed in primary tumors [76], which is not in agreement with the findings of Matsunoki et al. and our group. This discrepancy may be due to differences in the patient cohorts or methods used to assess the LINE-1 methylation levels or simply due to chance variation between independent studies. This issue must be further examined in independent studies with greater sample sizes in the future.
LINE-1 hypomethylation in colon cancer is recognized to be a prognostic biomarker. In a study of 643 colon cancers in two independent prospective cohorts, Ogino et al. demonstrated that the extent of LINE-1 hypomethylation is linearly associated with aggressive tumor behavior. They observed an approximately fivefold difference in cancer-specific mortality as the LINE-1 methylation levels of the tumor genes ranged from high to low [7]. Ahn et al. [77] showed that a lower LINE-1 methylation level is associated with an unfavorable prognosis in patients with resected stage III proximal, but not distal, colorectal cancer. The LINE-1 methylation level may also be a useful marker for predicting survival benefits from adjuvant chemotherapy. Surgery supplemented with adjuvant chemotherapy using oral fluoropyrimidines extends the lifespan of patients with low LINE-1 methylation levels, while apparently conferring no survival benefits in patients with high LINE-1 methylation levels [78]. These intriguing findings should be confirmed in future prospective studies.
The LINE-1 methylation level in normal colon tissue has also been explored. Kamiyama et al. [79] evaluated the LINE-1 methylation levels in matched cancer tissues and non-cancerous colonic mucosa in colorectal cancer patients and found a relationship between increased LINE-1 demethylation in the non-cancerous colonic mucosa and the presence of multiple (synchronous and metachronous) tumors. The LINE-1 methylation level in normal colonic mucosa is independent of age, sex, body mass index, smoking status, alcohol consumption and race [80]. Interestingly, the LINE-1 methylation levels in normal mucosa obtained from the right side of the bowel are significantly lower than those observed in mucosa obtained from the left side of the bowl [80]. This finding suggests that a field of predisposition toward colorectal cancer development arises in histologically normal colon mucosa (i.e. an epigenetic field defect or epigenetic field of cancer development). The LINE-1 methylation level is inversely associated with the CpG island methylation of the mlh1, p16, timp3, apc, er and myod genes, as well as the cimp gene in normal colonic mucosa [81]. Since folate plays a crucial role in methylation, its deficiency can contribute to DNA hypomethylation. Figueiredo et al. [80] reported that folic acid supplementation does not affect the LINE-1 methylation levels in normal colonic mucosa. In contrast, Liu et al. [82] demonstrated an association between the relative distribution of folate species and global DNA hypomethylation in normal human colorectal mucosa. Given that epigenetic alterations in normal mucosal tissues are emerging as a promising marker for cancer risk assessment [83–85], these findings are of considerable clinical relevance.
LINE-1 methylation in gastric cancer
Examining molecular changes is important for the development of innovative strategies for treating gastric cancer, especially those targeted to specific molecules [86]. Two large-scale (>200 cases) studies have examined how the LINE-1 methylation levels in patients with gastric cancer relate to the clinical outcomes. In a study by Bae et al. [87], the LINE-1 methylation level declined during progression from intestinal metaplasia to gastric adenoma, and then stabilized as gastric adenoma progressed to gastric cancer. The authors also found a strong correlation between LINE-1 hypomethylation and a poor prognosis in gastric cancer patients. At around the same time, our group reported that (1) the LINE-1 methylation levels are significantly lower in gastric cancer tissue than in matched normal gastric mucosa; (2) the tumoral LINE-1 methylation level ranges from 11.6 to 97.5 on a 0–100 scale (mean 71.4, median 74.4, SD 12.9); and (3) LINE-1 hypomethylation is correlated with shorter overall survival [88]. Collectively, LINE-1 hypomethylation in gastric cancer is associated with unfavorable clinical outcomes, suggesting its potential use as a prognostic biomarker.
Many studies have focused on the LINE-1 methylation levels in premalignant mucosa of the stomach. In a study by Yamamoto et al. [89], the LINE-1 methylation levels were significantly lower in the mucosal tissues obtained from patients with enlarged-fold gastritis than in healthy mucosae. The authors concluded that genome-wide hypomethylation occurs in enlarged-fold gastritis and may contribute to the tumorigenesis of diffuse-type gastric cancer. Park et al. [90] reported that the methylation of repetitive DNA elements (i.e. ALU and LINE-1) in gastric lesions generally decreases as the lesion progresses through the four stages from chronic to cancerous (chronic gastritis, intestinal metaplasia, gastric adenoma, gastric cancer). Although experienced pathologists have been unable to reach a consensus regarding the histological diagnosis of gastric intramucosal neoplastic lesions, the LINE-1 methylation level may function as a diagnostic tool for identifying high-grade dysplasia and intramucosal cancer. According to Lee et al. [91], the LINE-1 methylation level is a sensitive and specific tool for distinguishing high-grade dysplasia and intramucosal cancer from low-grade dysplasia. In gastric epithelial dysplasia resulting from Helicobacter pylori infection, MSI is correlated with a reduced LINE-1 methylation level, thus suggesting the coexistence of H. pylori and MSI as driving forces of gastric carcinogenesis [92]. Given that epigenetic damage induced by H. pylori may accumulate in gastric mucosae before malignancy develops [93], improving understanding of the relationship between H. Pylori infection and LINE-1 methylation may assist in estimating the risk of gastric cancer.
The association between the risk of cancer and the global DNA hypomethylation levels in blood leukocytes has been investigated in several types of cancers [94]. The LINE-1 methylation levels in peripheral blood leukocytes may function as a risk biomarker for head and neck cancer [39] and bladder cancer [51]. Two studies have evaluated the relationship between the LINE-1 methylation levels in peripheral blood leukocytes and the risk of gastric cancer. In a prospective cohort study of 192 gastric cancer patients and 384 matched controls, the risk of cancer was independent of the LINE-1 methylation level [95]. However, in a Polish population-based study of 302 gastric cancer patients and 421 age- and sex-matched controls, the risk of gastric cancer was highest among those with the lowest levels of LINE-1 methylation, although this trend was not statistically significant. Interestingly, subgroup analyses revealed stronger associations among individuals with a family history of cancer, current alcohol or tobacco users, those who rarely or never consumed fruit and CC and TT carriers of the MTRR Ex5+123C>T and MTRR Ex15+572T>C polymorphisms. These results suggest that blood leukocyte DNA hypomethylation and host behaviors potentially determine the risk of gastric cancer [96].
LINE-1 methylation in esophageal cancer
The impact of LINE-1 methylation on the development of esophageal cancer has received little attention. The majority of studies in this field have been published by our group [8, 35, 97]. First, we assessed the precision with which sodium bisulfite conversion and PCR-pyrosequencing assays can evaluate LINE-1 methylation in esophageal squamous cell carcinoma (ESCC) [35]. In order to measure the degree of assay precision, we performed bisulfite conversion on five different DNA specimen aliquots and subjected each specimen to five rounds of PCR pyrosequencing. Under different bisulfite treatments, we observed no substantial variation in the LINE-1 methylation levels and determined that a single bisulfite-treated DNA specimen can provide a precise measurement of LINE-1 methylation. The variation in the LINE-1 methylation levels between the PCR-pyrosequencing runs was similarly not large, and a single run of PCR pyrosequencing yielded a reasonably precise measurement of LINE-1 methylation in a given specimen. We also assessed the heterogeneity of the LINE-1 methylation levels in an esophageal tumor. To this end, we prepared five tissue sections obtained from a single tumor and examined the LINE-1 methylation level in each section (section-to-section). The LINE-1 methylation levels in the different tissue sections varied little, implying that the LINE-1 methylation level of a representative tissue section likely represents that of the whole tumor. Collectively, these findings suggest that the LINE-1 methylation level in ESCC can be reliably quantified using bisulfite-pyrosequencing assays. Second, using 217 curatively resected ESCC specimens, we evaluated the relationship between the LINE-1 methylation levels and patient prognoses [8]. The LINE-1 methylation levels were significantly lower in the ESCC mucosal tissues than in their matched normal counterparts. The level of LINE-1 hypomethylation in ESCC is associated with truncated survival, suggesting that, as for the other cancers discussed in this review, it may be used as a prognostic biomarker. Third, we proposed the potential of the LINE-1 methylation level as an indicator of the presence of an epigenetic field in ESCC cancerization [97]. Non-cancerous esophageal mucosa obtained from ESCC patients exhibits higher levels of LINE-1 methylation than similar tissues obtained from autopsied individuals without ESCC, suggesting that LINE-1 hypomethylation is a phenomenon of ESCC epigenetic field defects. In addition, LINE-1 hypomethylation is significantly associated with tobacco smoking, but not alcohol consumption. Our data support the use of the LINE-1 hypomethylation level as a surrogate marker of epigenetic field defects, particularly those caused by tobacco smoking.
Conclusions
LINE-1 is a repetitive DNA retrotransposon that duplicates via a copy-and-paste genetic mechanism. Importantly, because LINE-1 constitutes approximately 17 % of the human genome, the LINE-1 methylation level has been identified to be a surrogate marker of global DNA methylation. In this review, we summarized studies that have related the LINE-1 methylation levels to the patient prognosis, the presence of molecular alterations and clinical characteristics in subjects with GI cancers. In contrast to irreversible genetic changes, epigenetic changes may provide potentially reversible molecular targets for both cancer therapy and chemoprevention. Further investigations in this field would provide deeper insights into the pathogenesis of GI cancer and assist in the development of new therapeutic strategies against these cancers.
References
Taby R, Issa JP. Cancer epigenetics. CA Cancer J Clin. 2010;60(6):376–92.
Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–68.
Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17(3):330–9.
Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1(2):239–59.
Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, et al. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300(5618):489–92.
Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10(10):691–703.
Ogino S, Nosho K, Kirkner GJ, Kawasaki T, Chan AT, Schernhammer ES, et al. A cohort study of tumoral LINE-1 hypomethylation and prognosis in colon cancer. J Natl Cancer Inst. 2008;100(23):1734–8.
Iwagami S, Baba Y, Watanabe M, Shigaki H, Miyake K, Ishimoto T, et al. LINE-1 hypomethylation is associated with a poor prognosis among patients with curatively resected esophageal squamous cell carcinoma. Ann Surg. 2012;257(3):449–55.
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921.
de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011;7(12):e1002384.
Gemayel R, Vinces MD, Legendre M, Verstrepen KJ. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu Rev Genet. 2010;44:445–77.
Singer MF. SINEs and LINEs: highly repeated short and long interspersed sequences in mammalian genomes. Cell. 1982;28(3):433–4.
Penzkofer T, Dandekar T, Zemojtel T. L1Base: from functional annotation to prediction of active LINE-1 elements. Nucleic Acids Res. 2005;33((Database issue)):D498–500.
Ostertag EM, Kazazian HH Jr. Biology of mammalian L1 retrotransposons. Annu Rev Genet. 2001;35:501–38.
Babushok DV, Kazazian HH Jr. Progress in understanding the biology of the human mutagen LINE-1. Hum Mutat. 2007;28(6):527–39.
Iskow RC, McCabe MT, Mills RE, Torene S, Pittard WS, Neuwald AF, et al. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell. 2010;141(7):1253–61.
Schulz WA. L1 retrotransposons in human cancers. J Biomed Biotechnol. 2006;2006(1):83672.
Matlik K, Redik K, Speek M. L1 antisense promoter drives tissue-specific transcription of human genes. J Biomed Biotechnol. 2006;2006(1):71753.
Speek M. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Mol Cell Biol. 2001;21(6):1973–85.
Weber B, Kimhi S, Howard G, Eden A, Lyko F. Demethylation of a LINE-1 antisense promoter in the cMet locus impairs Met signalling through induction of illegitimate transcription. Oncogene. 2010;29(43):5775–84.
Swergold GD. Identification, characterization, and cell specificity of a human LINE-1 promoter. Mol Cell Biol. 1990;10(12):6718–29.
Thayer RE, Singer MF, Fanning TG. Undermethylation of specific LINE-1 sequences in human cells producing a LINE-1-encoded protein. Gene. 1993;133(2):273–7.
Hata K, Sakaki Y. Identification of critical CpG sites for repression of L1 transcription by DNA methylation. Gene. 1997;189(2):227–34.
Steinhoff C, Schulz WA. Transcriptional regulation of the human LINE-1 retrotransposon L1.2B. Mol Genet Genomics. 2003;270(5):394–402.
Chalitchagorn K, Shuangshoti S, Hourpai N, Kongruttanachok N, Tangkijvanich P, Thong-ngam D, et al. Distinctive pattern of LINE-1 methylation level in normal tissues and the association with carcinogenesis. Oncogene. 2004;23(54):8841–6.
Alves G, Tatro A, Fanning T. Differential methylation of human LINE-1 retrotransposons in malignant cells. Gene. 1996;176(1–2):39–44.
Phokaew C, Kowudtitham S, Subbalekha K, Shuangshoti S, Mutirangura A. LINE-1 methylation patterns of different loci in normal and cancerous cells. Nucleic Acids Res. 2008;36(17):5704–12.
Sepulveda AR, Jones D, Ogino S, Samowitz W, Gulley ML, Edwards R, et al. CpG methylation analysis—current status of clinical assays and potential applications in molecular diagnostics: a report of the Association for Molecular Pathology. J Mol Diagn. 2009;11(4):266–78.
Pobsook T, Subbalekha K, Sannikorn P, Mutirangura A. Improved measurement of LINE-1 sequence methylation for cancer detection. Clin Chim Acta. 2010;412(3–4):314–21.
Weisenberger DJ, Campan M, Long TI, Kim M, Woods C, Fiala E, et al. Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res. 2005;33(21):6823–36.
Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32(3):e38.
Vaissiere T, Cuenin C, Paliwal A, Vineis P, Hoek G, Krzyzanowski M, et al. Quantitative analysis of DNA methylation after whole bisulfitome amplification of a minute amount of DNA from body fluids. Epigenetics. 2009;4(4):221–30.
Aparicio A, North B, Barske L, Wang X, Bollati V, Weisenberger D, et al. LINE-1 methylation in plasma DNA as a biomarker of activity of DNA methylation inhibitors in patients with solid tumors. Epigenetics. 2009;4(3):176–84.
Irahara N, Nosho K, Baba Y, Shima K, Lindeman NI, Hazra A, et al. Precision of pyrosequencing assay to measure LINE-1 methylation in colon cancer, normal colonic mucosa, and peripheral blood cells. J Mol Diagn. 2010;12(2):177–83.
Iwagami S, Baba Y, Watanabe M, Shigaki H, Miyake K, Ida S, et al. Pyrosequencing assay to measure LINE-1 methylation level in esophageal squamous cell carcinoma. Ann Surg Oncol. 2012;19(8):2726–32.
Ahn JB, Chung WB, Maeda O, Shin SJ, Kim HS, Chung HC, et al. DNA methylation predicts recurrence from resected stage III proximal colon cancer. Cancer. 2010;117(9):1847–54.
Zhu ZZ, Hou L, Bollati V, Tarantini L, Marinelli B, Cantone L, et al. Predictors of global methylation levels in blood DNA of healthy subjects: a combined analysis. Int J Epidemiol. 2010;41(1):126–39.
Ibrahim AE, Arends MJ, Silva AL, Wyllie AH, Greger L, Ito Y, et al. Sequential DNA methylation changes are associated with DNMT3B overexpression in colorectal neoplastic progression. Gut. 2010;60(4):499–508.
Hsiung DT, Marsit CJ, Houseman EA, Eddy K, Furniss CS, McClean MD, et al. Global DNA methylation level in whole blood as a biomarker in head and neck squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. 2007;16(1):108–14.
Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301(5895):89–92.
Cho NY, Kim BH, Choi M, Yoo EJ, Moon KC, Cho YM, et al. Hypermethylation of CpG island loci and hypomethylation of LINE-1 and Alu repeats in prostate adenocarcinoma and their relationship to clinicopathological features. J Pathol. 2007;211(3):269–77.
Florl AR, Steinhoff C, Muller M, Seifert HH, Hader C, Engers R, et al. Coordinate hypermethylation at specific genes in prostate carcinoma precedes LINE-1 hypomethylation. Br J Cancer. 2004;91(5):985–94.
Yegnasubramanian S, Haffner MC, Zhang Y, Gurel B, Cornish TC, Wu Z, et al. DNA hypomethylation arises later in prostate cancer progression than CpG island hypermethylation and contributes to metastatic tumor heterogeneity. Cancer Res. 2008;68(21):8954–67.
Pattamadilok J, Huapai N, Rattanatanyong P, Vasurattana A, Triratanachat S, Tresukosol D, et al. LINE-1 hypomethylation level as a potential prognostic factor for epithelial ovarian cancer. Int J Gynecol Cancer. 2008;18(4):711–7.
Woloszynska-Read A, Mhawech-Fauceglia P, Yu J, Odunsi K, Karpf AR. Intertumor and intratumor NY-ESO-1 expression heterogeneity is associated with promoter-specific and global DNA methylation status in ovarian cancer. Clin Cancer Res. 2008;14(11):3283–90.
Furniss CS, Marsit CJ, Houseman EA, Eddy K, Kelsey KT. Line region hypomethylation is associated with lifestyle and differs by human papillomavirus status in head and neck squamous cell carcinomas. Cancer Epidemiol Biomarkers Prev. 2008;17(4):966–71.
Smith IM, Mydlarz WK, Mithani SK, Califano JA. DNA global hypomethylation in squamous cell head and neck cancer associated with smoking, alcohol consumption and stage. Int J Cancer. 2007;121(8):1724–8.
Daskalos A, Logotheti S, Markopoulou S, Xinarianos G, Gosney JR, Kastania AN, et al. Global DNA hypomethylation-induced DeltaNp73 transcriptional activation in non-small cell lung cancer. Cancer Lett. 2011;300(1):79–86.
Saito K, Kawakami K, Matsumoto I, Oda M, Watanabe G, Minamoto T. Long interspersed nuclear element 1 hypomethylation is a marker of poor prognosis in stage IA non-small cell lung cancer. Clin Cancer Res. 2011;16(8):2418–26.
Florl AR, Lower R, Schmitz-Drager BJ, Schulz WA. DNA methylation and expression of LINE-1 and HERV-K provirus sequences in urothelial and renal cell carcinomas. Br J Cancer. 1999;80(9):1312–21.
Wilhelm CS, Kelsey KT, Butler R, Plaza S, Gagne L, Zens MS, et al. Implications of LINE1 methylation for bladder cancer risk in women. Clin Cancer Res. 2010;16(5):1682–9.
Roman-Gomez J, Jimenez-Velasco A, Agirre X, Cervantes F, Sanchez J, Garate L, et al. Promoter hypomethylation of the LINE-1 retrotransposable elements activates sense/antisense transcription and marks the progression of chronic myeloid leukemia. Oncogene. 2005;24(48):7213–23.
Fabris S, Bollati V, Agnelli L, Morabito F, Motta V, Cutrona G, et al. Biological and clinical relevance of quantitative global methylation of repetitive DNA sequences in chronic lymphocytic leukemia. Epigenetics. 2011;6(2):188–94.
Subbalekha K, Pimkhaokham A, Pavasant P, Chindavijak S, Phokaew C, Shuangshoti S, et al. Detection of LINE-1s hypomethylation in oral rinses of oral squamous cell carcinoma patients. Oral Oncol. 2009;45(2):184–91.
Lee JJ, Geli J, Larsson C, Wallin G, Karimi M, Zedenius J, et al. Gene-specific promoter hypermethylation without global hypomethylation in follicular thyroid cancer. Int J Oncol. 2008;33(4):861–9.
Sunami E, Vu AT, Nguyen SL, Giuliano AE, Hoon DS. Quantification of LINE1 in circulating DNA as a molecular biomarker of breast cancer. Ann NY Acad Sci. 2008;1137:171–4.
Mirabello L, Savage SA, Korde L, Gadalla SM, Greene MH. LINE-1 methylation is inherited in familial testicular cancer kindreds. BMC Med Genet. 2010;11:77.
Estecio MR, Gharibyan V, Shen L, Ibrahim AE, Doshi K, He R, et al. LINE-1 hypomethylation in cancer is highly variable and inversely correlated with microsatellite instability. PLoS One. 2007;2(5):e399.
Daskalos A, Nikolaidis G, Xinarianos G, Savvari P, Cassidy A, Zakopoulou R, et al. Hypomethylation of retrotransposable elements correlates with genomic instability in non-small cell lung cancer. Int J Cancer. 2009;124(1):81–7.
Geisel J, Schorr H, Heine GH, Bodis M, Hubner U, Knapp JP, et al. Decreased p66Shc promoter methylation in patients with end-stage renal disease. Clin Chem Lab Med. 2007;45(12):1764–70.
Choi SH, Worswick S, Byun HM, Shear T, Soussa JC, Wolff EM, et al. Changes in DNA methylation of tandem DNA repeats are different from interspersed repeats in cancer. Int J Cancer. 2009;125(3):723–9.
Sigalotti L, Fratta E, Bidoli E, Covre A, Parisi G, Colizzi F, et al. Methylation levels of the “long interspersed nucleotide element-1” repetitive sequences predict survival of melanoma patients. J Transl Med. 2011;9:78.
Igarashi S, Suzuki H, Niinuma T, Shimizu H, Nojima M, Iwaki H, et al. A novel correlation between LINE-1 hypomethylation and the malignancy of gastrointestinal stromal tumors. Clin Cancer Res. 2010;16(21):5114–23.
Baer C, Claus R, Plass C. Genome-wide epigenetic regulation of miRNAs in cancer. Cancer Res. 2013;73(2):473–7.
Ogino S, Kawasaki T, Nosho K, Ohnishi M, Suemoto Y, Kirkner GJ, et al. LINE-1 hypomethylation is inversely associated with microsatellite instability and CpG island methylator phenotype in colorectal cancer. Int J Cancer. 2008;122(12):2767–73.
Markowitz SD, Bertagnolli MM. Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med. 2009;361(25):2449–60.
Goel A, Xicola RM, Nguyen TP, Doyle BJ, Sohn VR, Bandipalliam P, et al. Aberrant DNA methylation in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology. 2010;138(5):1854–62.
Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4(12):988–93.
Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA. 1999;96(15):8681–6.
Grady WM. CIMP and colon cancer gets more complicated. Gut. 2007;56(11):1498–500.
Baba Y, Huttenhower C, Nosho K, Tanaka N, Shima K, Hazra A, et al. Epigenomic diversity of colorectal cancer indicated by LINE-1 methylation in a database of 869 tumors. Mol Cancer. 2010;9:125.
Antelo M, Balaguer F, Shia J, Shen Y, Hur K, Moreira L, et al. A high degree of LINE-1 hypomethylation is a unique feature of early-onset colorectal cancer. PLoS One. 2012;7(9):e45357.
Sunami E, de Maat M, Vu A, Turner RR, Hoon DS. LINE-1 hypomethylation during primary colon cancer progression. PLoS One. 2011;6(4):e18884.
Matsunoki A, Kawakami K, Kotake M, Kaneko M, Kitamura H, Ooi A, et al. LINE-1 methylation shows little intra-patient heterogeneity in primary and synchronous metastatic colorectal cancer. BMC Cancer. 2012;12:574.
Murata A, Baba Y, Watanabe M, Shigaki H, Miyake K, Ishimoto T, et al. Methylation levels of LINE-1 in primary lesion and matched metastatic lesions of colorectal cancer. Br J Cancer. 2013;109:408–15.
Hur K, Cejas P, Feliu J, Moreno-Rubio J, Burgos E, Boland CR, et al. Hypomethylation of long interspersed nuclear element-1 (LINE-1) leads to activation of proto-oncogenes in human colorectal cancer metastasis. Gut. 2013 [Epub ahead of print].
Ahn JB, Chung WB, Maeda O, Shin SJ, Kim HS, Chung HC, et al. DNA methylation predicts recurrence from resected stage III proximal colon cancer. Cancer. 2011;117(9):1847–54.
Kawakami K, Matsunoki A, Kaneko M, Saito K, Watanabe G, Minamoto T. Long interspersed nuclear element-1 hypomethylation is a potential biomarker for the prediction of response to oral fluoropyrimidines in microsatellite stable and CpG island methylator phenotype-negative colorectal cancer. Cancer Sci. 2011;102(1):166–74.
Kamiyama H, Suzuki K, Maeda T, Koizumi K, Miyaki Y, Okada S, et al. DNA demethylation in normal colon tissue predicts predisposition to multiple cancers. Oncogene. 2012;31(48):5029–37.
Figueiredo JC, Grau MV, Wallace K, Levine AJ, Shen L, Hamdan R, et al. Global DNA hypomethylation (LINE-1) in the normal colon and lifestyle characteristics and dietary and genetic factors. Cancer Epidemiol Biomarkers Prev. 2009;18(4):1041–9.
Iacopetta B, Grieu F, Phillips M, Ruszkiewicz A, Moore J, Minamoto T, et al. Methylation levels of LINE-1 repeats and CpG island loci are inversely related in normal colonic mucosa. Cancer Sci. 2007;98(9):1454–60.
Liu J, Hesson LB, Meagher AP, Bourke MJ, Hawkins NJ, Rand KN, et al. Relative distribution of folate species is associated with global DNA methylation in human colorectal mucosa. Cancer Prev Res (Phila). 2012;5(7):921–9.
Chiba T, Marusawa H, Ushijima T. Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation. Gastroenterology. 2012;143(3):550–63.
Ushijima T, Asada K. Aberrant DNA methylation in contrast with mutations. Cancer Sci. 2010;101(2):300–5.
Ushijima T. Epigenetic field for cancerization. J Biochem Mol Biol. 2007;40(2):142–50.
Oshima T, Masuda M. Molecular targeted agents for gastric and gastroesophageal junction cancer. Surg Today. 2012;42(4):313–27.
Bae JM, Shin SH, Kwon HJ, Park SY, Kook MC, Kim YW, et al. ALU and LINE-1 hypomethylations in multistep gastric carcinogenesis and their prognostic implications. Int J Cancer. 2012;131(6):1323–31.
Shigaki H, Baba Y, Watanabe M, Murata A, Iwagami S, Miyake K, et al. LINE-1 hypomethylation in gastric cancer, detected by bisulfite pyrosequencing, is associated with poor prognosis. Gastric Cancer. 2013;16(4):480–7.
Yamamoto E, Toyota M, Suzuki H, Kondo Y, Sanomura T, Murayama Y, et al. LINE-1 hypomethylation is associated with increased CpG island methylation in Helicobacter pylori-related enlarged-fold gastritis. Cancer Epidemiol Biomarkers Prev. 2008;17(10):2555–64.
Park SY, Yoo EJ, Cho NY, Kim N, Kang GH. Comparison of CpG island hypermethylation and repetitive DNA hypomethylation in premalignant stages of gastric cancer, stratified for Helicobacter pylori infection. J Pathol. 2009;219(4):410–6.
Lee JR, Chung WC, Kim JD, Lee KM, Paik CN, Jung SH, et al. Differential LINE-1 hypomethylation of gastric low-grade dysplasia from high grade dysplasia and intramucosal cancer. Gut Liver. 2011;5(2):149–53.
Kim JS, Chung WC, Lee KM, Paik CN, Lee KS, Kim HJ, et al. Association between genetic instability and Helicobaaacter pylori infection in gastric epithelial dysplasia. Gastroenterol Res Pract. 2012;2012:360929.
Nanjo S, Asada K, Yamashita S, Nakajima T, Nakazawa K, Maekita T, et al. Identification of gastric cancer risk markers that are informative in individuals with past H. pylori infection. Gastric Cancer. 2012;15(4):382–8.
Woo HD, Kim J. Global DNA hypomethylation in peripheral blood leukocytes as a biomarker for cancer risk: a meta-analysis. PLoS One. 2012;7(4):e34615.
Gao Y, Baccarelli A, Shu XO, Ji BT, Yu K, Tarantini L, et al. Blood leukocyte Alu and LINE-1 methylation and gastric cancer risk in the Shanghai Women’s Health Study. Br J Cancer. 2012;106(3):585–91.
Hou L, Wang H, Sartori S, Gawron A, Lissowska J, Bollati V, et al. Blood leukocyte DNA hypomethylation and gastric cancer risk in a high-risk Polish population. Int J Cancer. 2010;127(8):1866–74.
Shigaki H, Baba Y, Watanabe M, Iwagami S, Miyake K, Ishimoto T, et al. LINE-1 hypomethylation in noncancerous esophageal mucosae is associated with smoking history. Ann Surg Oncol. 2012;19(13):4238–43.
Rhee YY, Kim MJ, Bae JM, Koh JM, Cho NY, Juhnn YS, et al. Clinical outcomes of patients with microsatellite-unstable colorectal carcinomas depend on L1 methylation level. Ann Surg Oncol. 2012;19(11):3441–8.
Hoshimoto S, Kuo CT, Chong KK, Takeshima TL, Takei Y, Li MW, et al. AIM1 and LINE-1 epigenetic aberrations in tumor and serum relate to melanoma progression and disease outcome. J Invest Dermatol. 2012;132(6):1689–97.
van Hoesel AQ, van de Velde CJ, Kuppen PJ, Liefers GJ, Putter H, Sato Y, et al. Hypomethylation of LINE-1 in primary tumor has poor prognosis in young breast cancer patients: a retrospective cohort study. Breast Cancer Res Treat. 2012;134(3):1103–14.
Ohka F, Natsume A, Motomura K, Kishida Y, Kondo Y, Abe T, et al. The global DNA methylation surrogate LINE-1 methylation is correlated with MGMT promoter methylation and is a better prognostic factor for glioma. PLoS One. 2011;6(8):e23332.
Aoki Y, Nojima M, Suzuki H, Yasui H, Maruyama R, Yamamoto E, et al. Genomic vulnerability to LINE-1 hypomethylation is a potential determinant of the clinicogenetic features of multiple myeloma. Genome Med. 2012;4(12):101.
Saito K, Kawakami K, Matsumoto I, Oda M, Watanabe G, Minamoto T. Long interspersed nuclear element 1 hypomethylation is a marker of poor prognosis in stage IA non-small cell lung cancer. Clin Cancer Res. 2010;16(8):2418–26.
Balassiano K, Lima S, Jenab M, Overvad K, Tjonneland A, Boutron-Ruault MC, et al. Aberrant DNA methylation of cancer-associated genes in gastric cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC-EURGAST). Cancer Lett. 2011;311(1):85–95.
Conflict of interest
The authors have no conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported in part by the Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Scientific Research, Grant Number 23689061.
Rights and permissions
About this article
Cite this article
Baba, Y., Murata, A., Watanabe, M. et al. Clinical implications of the LINE-1 methylation levels in patients with gastrointestinal cancer. Surg Today 44, 1807–1816 (2014). https://doi.org/10.1007/s00595-013-0763-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00595-013-0763-6