
Abstract
Aims
G-protein-coupled receptor 39 (GPR39) has been implicated in glucose homoeostasis, appetite control and gastrointestinal tract function.
Methods
This study used clonal BRIN-BD11 cells and mouse pancreatic islets to assess the insulin-releasing actions of trace metals believed to act via GPR39, and the second messenger pathways involved in mediating their effects. Micromolar concentrations of Zn2+, Cu2+, Ni2+ and Co2+ were examined under normoglycaemic and hyperglycaemic conditions. Mechanistic studies investigated changes of intracellular Ca2+, cAMP generation and assessment of cytotoxicity by LDH release. Cellular localisation of GPR39 was determined by double immunohistochemical staining.
Results
All trace metals (7.8–500 µmol/l) stimulated insulin release with Cu2+ being the most potent in isolated islets, with an EC50 value of 87 μmol/l. Zn2+ was the most selective with an EC50 value of 125 μmol/l. Enhancement of insulin secretion was also observed with Ni2+ (179 μmol/l) and Co2+ (190 μmol/l). These insulin-releasing effects were confirmed using clonal BRIN-BD11 cells which exhibited enhanced intracellular Ca2+ (p < 0.05–p < 0.001) and cAMP generation (p < 0.05–p < 0.001) in response to trace metals. Oral administration of Zn2+, Ni2+ and Cu2+ (50 µmol/kg together with 18 mmol/kg glucose) decreased the glycaemic excursion (p < 0.05–p < 0.01) and augmented insulin secretion (p < 0.05–p < 0.01) in NIH Swiss mice.
Conclusions
This study has demonstrated the presence of GPR39 and the insulinotropic actions of trace metals on BRIN-BD11 cells and pancreatic beta cells, together with their antihyperglycaemic actions in vivo. These data suggest that development of agonists capable of specifically activating GPR39 may be a useful new therapeutic approach for diabetes management.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
G-protein-coupled receptor 39 (GPR39) is a member of the ghrelin receptor family comprising of the ghrelin receptor, neurotensin receptors and motilin receptor that are activated by peptide hormones and neuropeptides [1]. GPR39 is constitutively active and an increase in GPR39 expression will increase intracellular signalling, without any agonist present [2]. Two isoforms of GPR39 exist, GPR39-1a is the full length active receptor, while GPR39-1b is the truncated form which is biologically inactive [3].
The human GPR39 is a single copy gene mapped to chromosome 2 at q21–q22, encoding the 453 amino acid GPR39 protein expressed in the pancreas, kidney, liver, adipose tissue, central nervous system and gastrointestinal tract [4–6]. A previous study identified GPR39 co-expression with insulin producing beta cells in pancreatic islets and in duct cells of the exocrine pancreas [7]. Interestingly, expression of GPR39 has been identified in human adipose tissue with decreased mRNA levels in obese and diabetic individuals [5]. Additionally, GPR39 expression was negatively correlated with blood glucose concentrations in obese type 2 diabetics, suggesting that GPR39 may have a role in insulin secretion [5]. Indeed, GPR39 has been implicated recently in glucose homoeostasis, control of appetite and gastrointestinal tract function with signalling to Gαq pathway with activation of phospholipase C and to Gαs with activation of adenylate cyclase and Gα 12/13 [6, 8].
Obestatin a 23 amino acid peptide was previously found to activate GPR39 which suppresses appetite, decreases weight gain and delays gastric emptying [9]. However, several studies have been unable to reproduce the finding that obestatin controls food intake and activates GPR39 [10–12]. Interestingly, extracellular zinc ions at micromolar concentrations stimulate GPR39 and this is of particular note in pancreatic islets where Zn2+ is stored and co-secreted in large amounts with insulin [13, 14]. In the endocrine pancreas, Zn2+ has a role in insulin packaging, secretion and signalling and also regulates glucagon secretion from adjacent pancreatic α-cells [15]. Zn2+ activation of GPR39 in the islets may have both autocrine and paracrine effects [16]. In cells overexpressing GPR39, Zn2+ has been shown to activate signal pathways including cAMP generation and elevation of intracellular Ca2+ [16]. A previous report found that metal ions including Zn2+ also have the ability to activate melanocortin-1 and melanocortin-4 receptors, indicating that zinc ions may not be fully selective for GPR39 [17]. Interestingly, in vivo overexpression of GPR39 in mice revealed a protective role against streptozotocin-induced beta cell failure [6] and treatment of mice with zinc sulphate in drinking water prevented multiple low-dose streptozotocin-induced diabetes [18]. Zinc deficiency in rats resulted in decreased insulin secretion and insulin resistance [19], whereas chronic zinc supplementation in ob/ob mice decreased hyperglycaemia which may be due to enhanced insulin action [20].
The effect of GPR39-deficiency in mice demonstrates the importance of this receptor in the endocrine pancreas, resulting in impaired glucose-stimulated insulin release, while islet architecture including beta cell mass remained normal [1, 7, 21]. Additionally, adult GPR39 knockout mice displayed increased body weight, body fat and alterations in both metabolic and gastrointestinal function when compared to wild-type mice [22]. Recent studies have established a link between diabetes, glucose intolerance and impaired cognition [23, 24]. A role for GPR39 in the hippocampus has been identified by Zn2+ addition enhancing intracellular Ca2+ which was blunted by both U73122 (PLC inhibitor) and YM-254890 (Gαq inhibitor) [25]. Interestingly, GPR39 has been identified as a novel inhibitor of apoptosis induced by oxidative and endoplasmic reticulum stress in a GPR39 overexpressing hippocampal cell line [26]. A recent study found that a zinc-deficient diet resulted in decreased GPR39 and BDNF protein expression in the frontal cortex in mice [27]. However, to date no studies in GPR39 knockout mice have identified any neural dysfunctions [16], while zinc transporter-3 (ZnT3)-deficient mice exhibited defects in cognitive behaviour, increased seizures and neurogenesis [28, 29]. In addition to zinc, other trace metals including Ni2+ have been implicated in activating GPR39 in a dose-dependent manner [10]. Conversely, a study by Sharir et al. [30] found no effects of Ni2+, Fe2+, Cu2+ or Pb2+ on intracellular Ca2+ in a GPR39-expressing keratinocyte cell line.
Very few studies have assessed the potential anti-diabetic effects of Zn2+ and other trace metals upon activation of GPR39. This study assessed GPR39 expression and localisation in islet cells and the ability Zn2+, Cu2+, Co2+ and Ni2+ to stimulate insulin release from clonal BRIN-BD11 cells, isolated islets and enhance glucose tolerance in mice.
Materials and methods
Materials
Collagenase (derived from Clostridium histolyticum), cAMP enzyme immunoassay kits, trace metal salts and most other reagents were obtained from Sigma-Aldrich (Poole, UK). RPMI-1640 media, foetal bovine serum, streptomycin, Hanks buffer, trypsin and penicillin were supplied by Gibco Life Technologies Ltd (Strathclyde, UK). CytoTox96 non-radioactive cytotoxicity assay kits and FLEX calcium assay reagent were purchased from Promega (Madison, WI, USA) and Molecular Devices (Sunnyvale, CA, USA), respectively. Rabbit anti-GPR39 antibody was purchased from Abcam (Cambridge, UK).
Insulin secretion
Generation and characterisation of the insulin-secreting BRIN-BD11 cells were outlined previously [31]. BRIN-BD11 cells were cultured with RPMI-1640 media (11.1 mM glucose) containing antibiotics (100U/ml penicillin and 0.1 mg/ml streptomycin) and 10 % foetal calf serum at 37 °C in an atmosphere of 95 % air and 5 % carbon dioxide. For acute insulin secretion studies, cells were detached using trypsin/EDTA and incubated overnight in 24-well plates with 150,000 cells per well. Cells were then pre-incubated for 40 min at 1.1 mmol/l glucose in Krebs buffer (KRBB comprising 4.7 mmol/l KCL, 115 mmol/l NaCl, 1.28 mmol/CaCl22H2O, 10 mmol/l NaHCO3, 5 g/l BSA, 1.2 mmol/l KH2PO4, 1.2 mmol/l MgSO47H2O pH 7.4). Test incubations were then performed at 37 °C for 20 min. ZnSO4, ZnCl2, NiSO4, CuCl2 and CoCl2 (7.8–500 μmol/l), plus GLP-1 at (10−7 mol/l) as positive control were tested at both 5.6 mmol/l and 16.7 mmol/l glucose, as indicated in the figures. Supernatants were removed, evaluated for lactate dehydrogenase (LDH) release as an indicator of cytotoxicity (as per manufacturer’s protocol) or frozen at −20 °C until determination of insulin by radioimmunoassay [32]. Cells exposed to 20 mM streptozotocin were used as positive control for cytotoxicity tests.
Pancreatic islets were isolated by collagenase digestion [33] from normal mice derived from the colony maintained at Aston University, UK [34]. After overnight culture as above, groups of 10 islets were incubated for 60 min at 37 °C in 1 ml of 1.1 mmol/l glucose KRBB. Test incubations were then carried out for a further 60 min at 11.1 mmol/l glucose with addition of various trace metals (7.8–500 μmol/l) as appropriate. Isolated pancreatic islets are glucose responsive [35] and 11.1 mM glucose used in this study is a stimulatory concentration. Insulin secretion and insulin content of islets, treated overnight with 1 ml acid ethanol, were determined by radioimmunoassay [32].
Intracellular Ca2+ and cAMP
For intracellular Ca2+ measurement, monolayers of BRIN-BD11 cells were seeded overnight, at a density of 80,000 cells per well in a 96-well black-walled clear bottom plate [36]. Cells were washed with 100 μl of KRBB and incubated for 60 min with Flex calcium assay kit reagent at 37 °C. ZnSO4, ZnCl2, NiSO4, CuCl2 and CoCl2 at 500 μmol/l were added at 5.6 mmol/l and 16.7 mmol/l glucose. Fluorometric data were obtained using the FLEX Station scanner at a wavelength of 525 nm (Molecular Devices). For cAMP determination, BRIN-BD11 cells were seeded in a 96-well plate at a density of 30,000 cells per well. Cells were washed with 300 μl KRBB for 40 min, and 150 μl of trace metals at 7.8–500 μmol/l was tested at 11.1 mol/l glucose. After 20 min, test solutions were removed and 0.1 M HCl (150 μl) was added to lyse the cells. Total cAMP production in the cell supernatants was measured using cAMP enzyme immunoassay kit according to the manufacturer’s protocol (Sigma, Poole, UK).
Histology
BRIN-BD11 cells were allowed to attach overnight to polylysine-coated slides and fixed using 4 % paraformaldehyde/PBS for 20 min. Antigen retrieval was achieved by incubation in sodium citrate (50 mmol/l) at 90 °C for 20 min. Pancreatic tissues from normal mice [34] were fixed in 4 % PFA/PBS, embedded in paraffin wax, and sections cut at 8 μm. Sections were mounted onto polylysine-coated slides and dried on a hot plate. Pancreatic sections were dewaxed and following antigen retrieval, slides were incubated overnight at 4 °C with guinea pig anti-insulin (1:500), guinea pig anti-glucagon (1:500) and rabbit anti-GPR39 (1:1000). After washing in PBS, sections were incubated with Alexa Fluor 488 fluorescein goat anti-rabbit or anti-guinea pig IgG and goat anti-guinea pig or anti-rabbit Alexa 594 nm IgG (1:400; Molecular Probes (Life Technologies Ltd, Paisley, UK)) for 45 min at 37 °C and DAPI nuclear stain for 15 min at 37 °C. Finally, slides were washed in PBS, mounted and analysed using a BX51 Olympus microscope equipped with an Olympus XM10 digital camera. Relative GPR39 quantification analysis was performed on BRIN-BD11 cells after exposure to ZnCl2, NiSO4, CuCl2 and CoCl2 at 500 µmol/l at 11.1 mmol/l glucose for 20 min. GPR39 and insulin immunofluorescence staining was performed as described above. Analysis was performed by Cell-F software (closed polygon icon), with >200 cells per treatment group. All slides were blinded, and a negative control slide was performed to ensure antibody specificity with omission of the primary antibody [37]. Cellular auto-fluorescence after exposure to metal ions was investigated to confirm no modulation of the fluorescence spectra by the cations. No changes in fluorescence intensity were observed (data not shown).
Analysis of GPR39 expression using western blot
BRIN BD11 cells were seeded at a density of 600,000 cells per well in 6-well plates and allowed to attach overnight. After 20 min acute exposure to 0.5 mM ZnCl2, NiSO4, CuCl2, CoCl2, total protein was extracted at 4 °C for 10 min using RIPA buffer containing 150 mM NaCl, 1.0 % Nonidet P-40, 0.5 % sodium deoxycholate, 0.1 % SDS, 50 mM Tris HCl, pH 7.6 and protease inhibitor cocktail. Protein concentration was determined using Bradford reagent (Sigma, UK). Equal amounts of protein were boiled at 95 °C with Laemmli buffer for 10 min (2 µg/µl final concentration), and sample (30 µg per well) was loaded on to pre-cast gels (NUPAGE 4–12 % Bis–Tris gels, Invitrogen, UK) and subjected to SDS-PAGE (100 V, 45 min). After transfer to nitrocellulose membrane for 2 h at 30 V, membranes were blocked with 5 % skimmed milk and probed with rabbit anti-GPR39 (1:300) (Abcam, UK)/mouse anti-ACTB (1:3000) (Abcam, UK). Membranes were probed with ECL horseradish peroxidase donkey anti-rabbit IgG/ECL horseradish peroxidase sheep anti-mouse IgG (1:10000) (GE Healthcare, UK) and detected using Luminata Forte HRP substrate (Millipore, UK). Data were normalised to ACTB and expressed relative to untreated control.
Acute effects of trace metals in vivo
Adult male (20–22 week) NIH Swiss mice (Harlan UK Ltd) were individually housed in an air-conditioned room at 22 ± 2 °C with 12-h light/12-h darkness cycles. Drinking water and standard rodent maintenance diet (Trouw Nutrition, Cheshire, UK) were supplied ad libitum. Non-fasted NIH Swiss mice (n = 6) received an oral injection of glucose alone (18 mmol/kg body weight) or in combination with trace metals (50 μmol/kg body weight). Blood samples were obtained from the cut tip from tail vein of conscious mice and centrifuged at 13,000 rpm for 3 min at 4 °C. Plasma glucose was measured by an automated glucose oxidase procedure using a Beckman glucose analyser and insulin determined by radioimmunoassay [32]. All animal experiments were carried out in accordance with the UK Animal (Scientific Procedures) Act 1986 and the ARRIVE guidelines for reporting experiments involving animals [38] and approved by the University of Ulster Animal Ethics Review Committee.
Statistics
Data are expressed as the mean ± the standard error of the mean (SEM). Results were compared using the Student’s t test or one-way ANOVA on Prism graph pad version 5.0. Differences in data were considered to be statistically significant for p < 0.05.
Results
Effects of trace metals on insulin secretion from BRIN-BD11 cells
Insulin secretory abilities of trace metals (ZnCl2, ZnSO4, CuCl2, NiSO4, CoCl2) at 7.8–500 μmol/l were determined using clonal BRIN-BD11 cells. At 5.6 mmol/l glucose, ZnCl2 at 15.7–500 μmol/l stimulated insulin secretion by 1.2- to 2.7-fold (p < 0.05–p < 0.001, EC50 219 μmol/l; Fig. 1a). ZnSO4 had similar potency with a 1.3- to 2.2-fold (p < 0.05–p < 0.001) increase in insulin release (EC50 250 μmol/l; Fig. 1b). CuCl2 was the most potent trace metal augmenting insulin secretion at 31.3–500 μmol/l (p < 0.001, EC50 104 μmol/l) with a 1.6- to 2.6-fold increase (Fig. 1c), whereas NiSO4 enhanced insulin secretion at 31.3–500 μmol/l by 1.3- to 1.9-fold (p < 0.05–p < 0.001, EC50 158 μmol/l; Fig. 1d). CoCl2 was the least potent trace metal tested, augmenting insulin release by 1.5- to 2.0-fold at 250–500 μmol/l (p < 0.05–p < 0.001, EC50 273 μmol/l; Fig. 1e). The EC50 values ranged from 273 μmol/l (CoCl2) to 104 μmol/l (CuCl2) at 5.6 mM glucose.

Effects of a ZnCl2 b ZnSO4 c CuCl2 d NiSO4 and e CoCl2 on insulin secretion from BRIN-BD11 cells at 5.6 mmol/l glucose. GLP-1 (10−7 mol/l) was used as positive control. Effects of f ZnCl2 on LDH release. Results are the mean ± SEM (n = 8) for insulin secretion and (n = 3) for LDH release. *p < 0.05, **p < 0.01 and ***p < 0.001 compared to glucose alone
At 16.7 mmol/l glucose, ZnCl2 at 31.3–500 μmol/l enhanced insulin secretion by 1.3- to 1.9-fold (p < 0.001, EC50 91 μmol/l; Fig. 2a), while ZnSO4 stimulated a 1.2- to 1.9-fold (p < 0.05–p < 0.001) increase in insulin release (EC50 82 μmol/l; Fig. 2b). CuCl2 augmented insulin secretion at 31.3–500 μmol/l (p < 0.05–p < 0.001, EC50 166 μmol/l) with a 1.3- to 2.1-fold increase (Fig. 2c), while NiSO4 stimulated insulin secretion at 62.5–500 μmol/l by 1.3- to 1.7-fold (p < 0.05–p < 0.001, EC50 125 μmol/l; Fig. 2d). Similar to 5.6 mmol/l glucose, CoCl2 was the least potent trace metal, augmenting insulin secretion by 1.3- to 1.4-fold at 250–500 μmol/l (p < 0.05–p < 0.01, EC50 185 μmol/l; Fig. 2e). The EC50 values ranged from 185 μmol/l (CoCl2) to 82 μmol/l (ZnSO4) at 16.7 mmol/l glucose. Comparison of EC50 values at 5.6 and 16.7 mmol/l glucose showed significantly lower values at 16.7 mmol/l glucose, indicating that the effects of trace metals on insulin release were glucose sensitive. None of the trace metals tested at 5.6 mmol/l or 16.7 mmol/l glucose affected LDH release, indicating no cytotoxicity see Figs. 1 and 2f for representative data.

Effects of a ZnCl2 b ZnSO4 c CuCl2 d NiSO4 and e CoCl2 on insulin secretion from BRIN-BD11 cells at 16.7 mmol/l glucose. GLP-1 (10−7 mol/l) used as positive control. Effects of f ZnCl2 on LDH release. Results are the mean ± SEM (n = 8) for insulin secretion and (n = 3) for LDH release. *p < 0.05, **p < 0.01 and ***p < 0.001, compared to glucose alone
Effects of trace metals on intracellular Ca2+ and cAMP in BRIN-BD11 cells
Exposure of BRIN-BD11 cells to trace metals (ZnCl2, ZnSO4, CuCl2, NiSO4, CoCl2) at 500 μmol/l and alanine at 10 mmol/l resulted in potent stimulatory effects on intracellular Ca2+ at both 5.6 mmol/l and 16.7 mmol/l glucose (except CoCl2 at 16.7 mM; p < 0.05–p < 0.001; Fig. 3). Dose-dependent increase in cAMP production in BRIN-BD11 cells was observed with each GPR39 agonist and with GLP-1 at 10−7 mol/l. The agonists ZnCl2, CuCl2 and NiSO4 at 31.3–500 μmol/l increased cAMP production (p < 0.05–p < 0.001; Fig. 4a, c, d). ZnSO4 moderately increased cAMP production at 125–500 μmol/l (p < 0.05–p < 0.01; Fig. 4b), while CoCl2 was the least potent enhancing cAMP production only at 500 μmol/l (p < 0.05; Fig. 4e).

Effects of various trace metals (500 μmol/l) and alanine (10 mmol/l) on intracellular Ca2+ in BRIN-BD11 cells at 5.6 or 16.7 mmol/l glucose expressed as a, b RFU c, d Area under the curve. Results are the mean ± SEM (n = 8). *p < 0.05, **p < 0.01 and ***p < 0.001 compared to control

Effects of a ZnCl2 b ZnSO4 c CuCl2 d NiSO4 and e CoCl2 on cAMP production in BRIN-BD11 cells at 11.1 mmol/l glucose. GLP-1 (10−7 mol/l) used as positive control. Values are the mean ± SEM (n = 3). *p < 0.05, **p < 0.01 and ***p < 0.001 compared to 11.1 mmol/l glucose control
Effects of trace metals on insulin secretion from isolated islets
As displayed in Fig. 5, trace metal agonists had similar potencies on insulin secretion when tested in isolated islets. At 11.1 mM glucose, ZnCl2 at 15.7–500 μmol/l (p < 0.05–p < 0.001, EC50 125 μmol/l) enhanced insulin secretion (Fig. 5a), while ZnSO4 augmented insulin secretion at 31.3–500 μmol/l (p < 0.01–p < 0.001, EC50 125 μmol/l; Fig. 5b). CuCl2 was the most potent GPR39 agonist, increasing insulin release at 15.7–500 μmol/l (p < 0.05–p < 0.001, EC50 87 μmol/l; Fig. 5c), while NiSO4 enhanced insulin secretion at 31.3–500 μmol/l (p < 0.05–p < 0.001, EC50 179 μmol/l; Fig. 5d). The least potent agonist tested was CoCl2, augmenting insulin secretion at 125–500 μmol/l (p < 0.05–p < 0.001, EC50 190 μmol/l; Fig. 5e).

Effects of a ZnCl2 b ZnSO4 c CuCl2 d NiSO4 and e CoCl2 f all trace metals tested, with EC50 values on insulin release (% of insulin content) from isolated mouse islets at 11.1 mmol/l glucose. GLP-1 (10−7 mol/l) used as the positive control. Values are the mean ± SEM (n = 3). *p < 0.05, **p < 0.01 and ***p < 0.001, compared to 11.1 mmol/l glucose control
Expression of GPR39 in BRIN-BD11 cells and mouse islets
Zn, Ni, Cu and Co serve as GPR39 agonists in many cellular systems. The distribution of GPR39 and insulin in BRIN-BD11 cells was displayed in Fig. 6. Nuclei were stained by DAPI (blue) (Fig. 6a), and insulin (green) was located throughout the cells (Fig. 6b), with a similar staining pattern to GPR39 (red) (Fig. 6c). Double immunofluorescence of insulin with GPR39 indicated areas of co-localisation in BRIN-BD11 cells (yellow) (Fig. 6d). Selectivity of the various trace metals at 500 μmol/l was determined by relative fluorescence intensity at 11.1 mM glucose (Fig. 7a). ZnCl2 was highly specific for GPR39 increasing expression of GPR39 (red) (p < 0.001, Fig. 7b, g), while both CuCl2 and NiSO4 were moderately specific for GPR39 (p < 0.01, Fig. 7c, d, g). The least selective agonist tested was CoCl2 increasing expression of GPR39 (p < 0.05, Fig. 7e, g), while none of the agonists enhanced relative insulin intensity (green) in BRIN-BD11 cells (Fig. 7f, g).

Distribution of a DAPI nuclear stain, b insulin c GPR39 d merge of GPR39 co-localised with insulin at ×40 magnification in BRIN-BD11 cells. Examples of co-localisation indicated by arrows

Relative GPR39 fluorescence intensity of BRIN-BD11 cells following exposure to trace metals at 500 µmol/l at 11.1 mmol/l glucose for 20 min. GPR39 intensity in the presence of a 11.1 mmol/l glucose b ZnCl2, c CuCl2, d NiSO4, e CoCl2. f Representative image for insulin and g Relative GPR39 fluorescence intensity. Values are the mean ± SEM for > 200 cells per group determined by Cell-F software. *p < 0.05, **p < 0.01, ***p < 0.001 compared to 11.1 mmol/l glucose
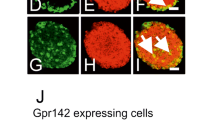
Figure 8 displays the localisation of insulin, glucagon and GPR39 in mouse pancreatic islets. DAPI (blue) stained the nuclei in pancreatic islets (Fig. 8a, b). GPR39 (red) was distributed throughout the islet (Fig. 8c, d), displaying a similar staining pattern to insulin (green) (Fig. 8e). Combination of insulin with GPR39 indicated that insulin-secreting beta cells express GPR39 (yellow) (Fig. 8g), examples of co-localisation are shown by the arrows. Glucagon (green) was located at the periphery of the islets (Fig. 8f) with no areas of co-localisation observed with GPR39 (Fig. 8h).

Distribution in mouse islets and pancreas a, b DAPI nuclear stain, c, d GPR39 e insulin f glucagon g merge of GPR39 co-localised with insulin. Example of co-localisation indicated by arrows. h merge of GPR39 and glucagon in mouse pancreatic tissue at ×40 magnification
Up-regulation of GPR39 protein expression (Fig. 7g) was confirmed by western blot studies (Fig. 9). All trace metals increased GPR39 protein expression in pancreatic BRIN-BD11 cells (Fig. 9), with significant effects being observed with Zn2+ (p < 0001), Ni2+ (p < 0.01) and Co2+ (p < 0.01).

a GPR39 and ACTB expression in control (lanes 1–3), ZnCl2 (lanes 4–6) and NiSO4 (lanes 7–9). b GPR39 and ACTB expression in control (lanes 1–3), CuCl2 (lanes 4–6) and CoCl2 (lanes 7–9). c, d Relative density (%), with GPR39 expression normalised to ACTB expression. Protein expression was determined in BRIN-BD11 cells after 20 min acute exposure to 0.5 mM trace metals. Values are mean ± SEM (n = 3). **p < 0.01, ***p < 0.001 compared to control (11.1 mM glucose)
Acute effects of trace metals on glucose tolerance and insulin release in vivo
An oral glucose tolerance test was utilised to determine the glucose-lowering and anti-diabetic potential of ZnCl2, CuCl2, NiSO4 and CoCl2 in vivo at 50 μmol/l in non-fasted NIH Swiss mice (Fig. 10a). Both ZnCl2 and CuCl2 reduced plasma glucose by 27 and 23 % (p < 0.05), respectively, after 60 min when compared to glucose (Fig. 10a). ZnCl2 (30 %, p < 0.05), CuCl2 (23 %, p < 0.01) and NiSO4 (15 %, p < 0.01) reduced the glycaemic excursion after 105 min (Fig. 10a). This reduction in glucose was confirmed by AUC values for ZnCl2 (p < 0.01), CuCl2 (p < 0.01) and NiSO4 (p < 0.05; Fig. 10c). Trace metals enhanced glucose-induced insulin secretion. Stimulatory effects of ZnCl2 (38 %, p < 0.01), CuCl2 (35 %, p < 0.01) and NiSO4 (33 %, p < 0.05) were observed at 30 min and effects of ZnCl2 (44 %, p < 0.01), CuCl2 (40 %, p < 0.01) persisted to 60 min (Fig. 10b). All trace metals assayed with exception of CoCl2 augmented insulin secretion (p < 0.05–p < 0.01) when compared to glucose alone (Fig. 10d).

Acute effects of trace metals on a oral glucose tolerance b plasma insulin response to glucose. AUC values for 0–105 min post injection for c plasma glucose and d plasma insulin are shown. Glucose (18 mmol/kg body weight) in combination with agonists (50 μmol/kg body weight) was administered by oral injection to non-fasted NIH Swiss mice (n = 6). Values are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 compared to glucose alone
Discussion
Interest in islet cell GPRs as a possible drug therapy for diabetes has intensified owing to successful exploitation of GLP-1 receptor mimetics and their general ability to counteract defective insulin secretion and beta cell loss [39]. Interestingly, several GPRs have been identified on enteroendocrine cells in the gastrointestinal tract and when activated enhance GLP-1 or GIP secretion, potentially increasing insulin release and beta cell regeneration [40]. Currently, a GPR40 (FFAR1) oral agonist TAK-875 is undergoing phase III clinical trials and has been shown to decrease glucose and HbA1C with equal potency to the sulphonylurea glimepiride but without the risk of hypoglycaemia [41]. GPR39 agonists may also stimulate insulin secretion and inhibit beta cell apoptosis which could be useful in the treatment of type 1 and 2 diabetes.
The current study investigated the insulin secretory effects of trace metals believed to act via GPR39 using clonal BRIN-BD11 cells, isolated mouse islets and NIH Swiss mice. Previous studies on various aspects of cellular metabolism using GPR39 knockout mice, siRNA knockdown and chemical antagonists have established their actions at GPR39 [10, 14, 27, 42]. Micromolar amounts of trace metals ZnCl2, ZnSO4, CuCl2, NiSO4 and CoCl2 enhanced insulin release in a concentration-dependent manner from clonal BRIN-BD11 cells and isolated islets under both normoglycaemic and hyperglycaemic conditions. At 5.6 mM glucose, CuCl2 was the most potent GPR39 agonist according to EC50 values, followed by NiSO4, ZnCl2, ZnSO4 and CoCl2. While at 16.7 mM glucose, ZnSO4 was the most potent, followed by ZnCl2, NiSO4, CuCl2 and CoCl2. These data are consistent with previous studies, indicating the potential role of GPR39 agonists in insulin secretion [7, 21, 43, 44]. Cytotoxicity testing measured by cellular LDH release indicated that none of the trace metals tested had adverse effects at the concentrations employed. Similarly others have reported lack of cytotoxicity of ZnSO4 at micromolar concentrations in a human intestinal epithelial cell line [45]. Intriguingly, administration of zinc sulphate to mice prevented multiple low-dose streptozotocin-induced diabetes and consequently, GPR39 has been described as a novel inhibitor of apoptosis [18].
Isolated mouse islets were used to substantiate findings made with clonal beta cells. Reassuringly, trace metals had similar insulin-releasing potency in order of increasing EC50 values namely CuCl2, ZnCl2, ZnSO4, NiSO4 and CoCl2. Inhibitory effects of trace metals such as Zn2+ on glucose-stimulated insulin release from mouse islets may occur independently of GPR39 [43]. However, such negative effects due to inhibition of Ca2+ influx transport are generally observed at millimolar concentrations [46]. In the present study, the mode of action of micromolar concentrations of trace metals was examined by measuring changes of intracellular Ca2+ and cAMP production. ZnCl2, ZnSO4, NiSO4 and CuCl2 each augmented intracellular Ca2+, demonstrating the stimulatory effects are mediated in large part through Ca2+-dependent pathways. Compared with GLP-1, trace metals also caused a moderate enhancement in total cAMP accumulation, with maximum effects being displayed with Zn2+, Ni2+ and Cu2+. In previous studies, exogenous Zn2+ at micromolar concentrations was found to activate K+ ATP channels in clonal beta cells, indicating a possible role in regulation of beta cell function [47], while activation of GPR39 has been linked to increases in cAMP accumulation and IP turnover [6, 8, 16]. The mechanism of GPR39 ligand-induced insulin secretion seems to predominately involve the Ca2+-dependent pathway and to a lesser extent the adenylyl cyclase pathway.
Evaluation of cellular localisation of GPR39 in BRIN-BD11 cells and pancreatic islets revealed GPR39 co-localisation with insulin in both clonal and primary beta cells. While no co-localisation was evident with glucagon on alpha cells, GPR39-mediated effects on pancreatic beta cells may exert a paracrine effect on other islet cell types. Previous studies have indicated GPR39 expression in mouse clonal beta cells [43], pancreatic beta cells and pancreatic duct epithelium [7]. Acute incubation of trace metals with BRIN-BD11 cells and quantification of GPR39 protein expression provided valuable information on agonist specificity for the receptor in this study. Zn2+ was highly specific for GPR39, augmenting receptor expression in islets, followed in potency by Cu2+, Ni2+ and lastly Co2+.
Trace metals assessed in this study are not totally specific for GPR39 and have the ability to interfere with ion channels and activate other receptors including melanocortin-1, melanocortin-4 receptors [17] and P2X4 receptor [48]. Although these trace metals have the ability to activate other receptors, previous studies have shown Zn2+ [6, 14, 16] and Ni2+ [10] to have high affinity for GPR39. Further, all trace metals examined are essential trace elements with diverse beneficial actions within the body [49]. Our acute in vivo data revealed that oral administration of these trace metals in particular Zn2+, Cu2+ and Ni2+ improved glucose tolerance and enhanced glucose-induced insulin release. These effects are presumably mediated by direct stimulation of insulin secretion by the trace metals, triggered via GPR39 combined with the possible secretion of GLP-1 and GIP from GPR39-expressing enteroendocrine cells. Indeed, GPCRs including free fatty acid receptors GPR119 and GPR120 have been implicated in the secretion of GLP-1, GIP and CCK on intestinal L, K and I cells and the secretion of these insulinotropic hormones [50, 51]. Additional actions of trace metals such as Zn2+ and Cu2+ might include stimulation of cellular glucose update and effects on multiple tissues due to modulation of Ca2+ fluxes. Further studies are necessary to evaluate effects of GPR39 agonists. Recently, oral administration of a Zn compound in a type 2 diabetes mouse model decreased blood glucose, indicating that Zn2+ may be utilised as an oral therapeutic agent for treatment of diabetes [52].
In conclusion, trace metal ligands targeting GPR39 on beta cells augment insulin secretion by increasing intracellular Ca2+ and cAMP generation. The acute in vivo glucose-lowering actions of these agonists indicate that activation of GPR39 with natural or synthetic metal ions agonists may provide new future therapies for diabetes. Further studies using specific siRNA for gene knockdown studies or genetic deletion in GPR39 knockout mice are required to fully understand the therapeutic effectiveness of GPR39.
References
Tremblay F, Perreault M, Klaman LD, Tobin JE, Smith E, Gimeno RE (2007) Normal food intake and body weight in mice lacking the G protein-coupled receptor GPR39. Endocrinology 148:501–506
Holst B, Holliday ND, Bach A, Elling CE, Cox HM, Schwartz TM (2004) Common structural basis for constitutive activity of the ghrelin receptor family. J Biol Chem 279:53806–53817
Egerod KL, Holst B, Petersen PS, Hansen JB, Mulder J, Hokfelt T, Schwartz TW (2007) GPR39 splice variants versus antisense gene LYPD1: expression and regulation in gastrointestinal tract, endocrine pancreas, liver and white adipose tissue. Mol Endocrinol 21:1685–1698
McKee KK, Tan CP, Palyha OC, Liu J, Feighner SD, Hreniuk DL, Smith RG, Howard AD, Van der Ploeg LH (1997) Cloning and characterization of two human G protein-coupled receptor genes (GPR38 and GPR39) related to growth hormone secretagogue and neurotensin receptors. Genomics 46:426–434
Catalan V, Gomez-Ambrosi J, Rotellar F, Silva C, Gil MJ, Rodriquez A, Cienfuegos JA, Salvador J, Fruhbeck G (2007) The obestatin receptor (GPR39) is expressed in human adipose tissue and is down-regulated in obesity-associated type 2 diabetes mellitus. Clin Endocrinol 66:598–601
Egerod KL, Jin C, Petersen PS, Wierup N, Sundler F, Holst B, Schwartz TW (2011) β- cell specific overexpression of GPR39 protects against streptozotocin-induced hyperglycaemia. Int J Endocrinol 2011:1–8
Holst B, Egerod KL, Jin C, Petersen PS, Ostergaard MV, Hald J, Sprinkel AM, Storling J, Mandrup-Poulsen T, Holst JJ et al (2009) G protein-coupled receptor 39 deficiency is associated with pancreatic islet dysfunction. Endocrinology 150:2577–2585
Xie F, Lui H, Zhu Y, Qin YR, Dai Y, Zeng T, Chen L, Nie C, Tang H, Li Y et al (2011) Overexpression of GPR39 contributes to malignant development of human esophageal squamous cell carcinoma. BMC Cancer 11:1–12
Zhang JV, Ren P, Avsian-Kretchmer O, Luo CW, Rauch R, Klein C, Hsueh AJ (2005) Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin’s effects on food intake. Science 310:996–999
Holst B, Egerod KL, Schild E, Vickers SP, Cheetham S, Gerlach LO, Storjohann L, Stidsen CE, Jones R, Beck-Sickinger AG et al (2007) GPR39 signaling is stimulated by zinc ions but not by obestatin. Endocrinology 148:13–20
Gourcerol G, St-Pierre DH, Tache Y (2007) Lack of obestatin effects on food intake: should obestatin be renamed ghrelin-associated peptide (GAP)? Regul Pept 141:1–7
Kobelt P, Wisser AS, Stengel A, Goebel M, Bannert N, Gourcerol G, Inhoff T, Noetzel S, Wiedenmann B, Klapp BF et al (2008) Peripheral obestatin has no effects on feeding behaviour and brain Fos expression in rodents. Peptides 29:1018–1027
Andersson T, Berggren PO, Flatt PR (1980) Subcellular distribution of zinc in islet β-cell fractions. Horm Metab Res 12:275–276
Petersen PS, Jin C, Madsen AN, Rasmussen M, Kuhre R, Egerod KL, Nielsen LB, Schwartz TW, Holst B (2011) Deficiency of the GPR39 receptor is associated with obesity and altered adipocyte metabolism. FASEB J 25:3803–3814
Kelleher SL, McCormick NH, Velasquez V, Lopez V (2011) Zinc in specialized secretory tissues: roles in the pancreas, prostate, and mammary gland. Adv Nutr 2:101–111
Popovics P, Stewart AJ (2011) GPR39: a Zn2+ -activated G-protein coupled receptor that regulates pancreatic, gastrointestinal and neuronal functions. Cell Mol Life Sci 68:85–95
Holst B, Elling CE, Schwartz TW (2002) Metal ion-mediated agonism and agonist enhancement in melanocortin MC1 and MC4 receptors. J Biol Chem 277:47662–47670
Ohly P, Dohle C, Abel J, Seissler J, Gleichmann H (2000) Zinc sulphate induces metallothionein in pancreatic islets of mice and protects against diabetes induced by multiple low doses of streptozotocin. Diabetologia 43:1020–1030
Huber AM, Gershoff SN (1973) Effect of zinc deficiency in rats on insulin release from the pancreas. J Nutr 103:1739–1744
Chen MD, Liou SJ, Lin PY, Yang VC, Alexander PS, Lin WH (1998) Effects of zinc supplementation on the plasma glucose level and insulin activity in genetically obese (ob/ob) mice. Bio Trace Elem Res 61:303–311
Depoortere I (2012) GI functions of GPR39: novel biology. Curr Opin Pharmacol 12:647–652
Moechars D, Depoortere I, Moreaux B, de Smet B, Goris I, Hoskens L, Daneels G, Kass S, Ver Donck L, Peeters T et al (2006) Altered gastrointestinal and metabolic function in the GPR39-obestatin receptor knockout mouse. Gastroenterology 131:1131–1141
Gault VA, Porter WD, Flatt PR, Holscher C (2010) Actions of exendin-4 therapy on cognitive function and hippocampal synaptic plasticity in mice fed a high fat diet. In J Obes 34:1341–1344
Porter DW, Kerr BD, Flatt PR, Holscher C, Gault VA (2010) Four weeks administration of Liraglutide improves memory and learning as well as glycaemic control in mice with dietary-induced obesity and insulin resistance. Diabetes Obes Metab 12:891–899
Besser L, Chorin E, Sekler I, Silverman WF, Atkin S, Russell J, Hershfinkel M (2009) Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J Neurosci 29:2890–2901
Dittmer S, Sahin M, Pantlen A, Saxena A, Toutzaris D, Pina AL, Geerts A, Golz S, Methner A (2008) The constitutively active orphan G-protein-coupled receptor GPR39 protects from cell death by increasing secretion of pigment epithelium-derived growth factor. J Biol Chem 283:7074–7081
Mlyneic K, Budziszewska B, Reczynski W, Sowa-Kucma M, Nowak G (2013) The role of the GPR39 receptor in zinc deficient-animal model of depression. Behav Brain Res 238:30–35
Cole TB, Robbins CA, Wenzel HJ, Schwartzkroin PA, Palmiter RD (2000) Seizures and neuronal damage in mice lacking vesicular zinc. Epilepsy Res 39:153–169
Sun SW, Won SJ, Hamby AM, Yoo BH, Fan Y, Sheline CT, Tamano H, Takeda A, Liu J (2009) Decreased brain zinc availability reduced hippocampal neurogenesis in mice and rats. J Cereb Blood Flow Metab 29:1579–1588
Sharir H, Zinger A, Nevo N, Sekler I, Hershfinkel M (2010) Zinc released from injured cells is acting via the Zn2+-sensing receptor, ZnR, to trigger signaling leading to epithelial repair. J Biol Chem 285:26097–26106
McClenaghan NH, Barnett CR, Ah-Sing E, Abdel-Wahab YH, O’Harte FP, Yoon TW, Swanston-Flatt SK, Flatt PR (1996) Characterisation of a novel glucose-responsive insulin-secreting cell line, BRIN BD11, produced by electrofusion. Diabetes 45:1132–1140
Flatt PR, Bailey CJ (1981) Abnormal plasma glucose and insulin responses in heterozygous lean (ob/+) mice. Diabetologia 20:573–577
Moskalewski S (1969) Studies on the culture and transplantation of isolated islets of Langerhans of the guinea pig. Proc K Ned Akad Wet C 72:157–171
Bailey CJ, Flatt PR (1982) Influence of genetic background and age on the expression of the obese hyperglycaemic syndrome in Aston ob/ob mice. Int J Obes 6:11–21
Hannan JMA, Marenah L, Ali L, Rokeya B, Flatt PR, Abdel-Wahab YHA (2006) Ocimum sanctum leaf extracts stimulate insulin secretion from perfused pancreas, isolated islets and clonal pancreatic β-cells. J Endocrinol 189:127–136
Miguel JC, Patterson S, Abdel-Wahab YHA, Mathias PC, Flatt PR (2004) Time-correlation between membrane depolarization and intracellular calcium in insulin secretion BRIN-BD11 cells: studies using FLIPR. Cell Calcium 36:43–50
Moran BM, Abdel-Wahab YH, Flatt PR, McKillop AM (2014) Evaluation of the insulin releasing and glucose lowering effects of GPR120 activation in pancreatic beta cells. Diabetes Obes Metab 16:1128–1139
Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010) Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160:1577–1579
Vangaveti V, Shashidhar V, Jarrod G, Baune BT, Kennedy RL (2010) Free fatty acid receptors: emerging targets for treatment of diabetes and its complications. Ther Adv Endocrinol Metab 1:165–175
Bailey CJ (2005) Drugs on the horizon for diabesity. Curr Diabet Rep 5:353–359
Burant CF (2013) Activation of GPR40 as a therapeutic target for the treatment of type 2 diabetes. Diabetes Care 36:S175–S179
Cohen L, Azriel-Tamir H, Arotsker N, Sekler I, Hershfinkel M (2012) Zinc sensing receptor signalling, mediated by GPR39, reduces butyrate-induced cell death in HT29 colonocytes via upregulation of clusterin. PloS One 7:e35482
Tremblay F, Richard A, Will S, Syed J, Stedman N, Perreault M, Gimeno RE (2009) Distribution of G-protein-coupled receptor 39 impairs insulin secretion in vivo. Endocrinology 150:2586–2595
Rutter GA (2010) Think zinc: new roles for zinc in the control of insulin secretion. Islets 2:49–50
Lodemann U, Einspanier R, Scharfen F, Martens H, Bondzio A (2013) Effects of zinc on epithelial barrier properties and viability in a human and a porcine intestinal cell culture model. Toxicol In Vitro 27:834–843
Flatt PR, Rorsman P, Swanston-Flatt SK (1987) Effect of cationic modifications on superficial binding and intracellular 45Ca uptake by decapsulated ob/ob mouse pancreatic islets. Biomed Res 8:153–159
Bloc A, Cens T, Cruz H, Dunant Y (2000) Zinc-induced changes in ionic currents of clonal rat pancreatic β-cells: activation of ATP-sensitive K+ channels. J Physiol 529:723–734
Acuna-Castillo C, Morales B, Huidobro-Toro JP (2000) Zinc and copper modulate differentially the P2X4 receptor. J Neurosci 74:1529–1537
Fraga CG (2005) Relevance, essentiality and toxicity of trace elements in human health. Mol Asp Med 26:235–244
Tanaka T, Yano T, Adachi T, Koshimizu TA, Hirasawa A, Tsujimoto G (2008) Cloning and characterization of the rat free fatty acid receptor GPR120: in vivo effect of the natural ligand on GLP-1 secretion and proliferation of pancreatic beta cells. N-S Arch Pharmacol 377:515–522
Lauffer LM, Iakoubov R, Brubaker PL (2009) GPR119 is essential for oleoylethanolamide–induced glucagon-like peptide-1 secretion from the intestinal enteroendocrine L-cells. Diabetes 58:1058–1066
Adachi Y, Yoshida J, Kodera Y, Kiss T, Jakusch T, Enyedy EA, Yoshikawa Y, Sakurai H (2006) Oral administration of a zinc complex improves type 2 diabetes and metabolic syndromes. Biochem Biophys Res Commun 351:165–170
Acknowledgments
These studies were supported by the Department of Education and Learning, Northern Ireland.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standard
This study was approved by the University of Ulster Animal Ethics Review Committee. All animal experiments were carried out in accordance with the UK Animal (Scientific Procedures) Act 1986.
Human and animal rights
All procedures followed were in accordance with the UK Animal (Scientific Procedures) Act 1986 and the ARRIVE guidelines for reporting experiments involving animals. No clinical studies were carried out in this study.
Informed consent
No informed consent was required as no patients or clinical studies were involved in this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Managed by Massimo Porta.
Rights and permissions
About this article

Cite this article
Moran, B.M., Abdel-Wahab, Y.H.A., Vasu, S. et al. GPR39 receptors and actions of trace metals on pancreatic beta cell function and glucose homoeostasis. Acta Diabetol 53, 279–293 (2016). https://doi.org/10.1007/s00592-015-0781-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00592-015-0781-5