
Abstract
Understanding of the ecology of arbuscular mycorrhizal fungi comes primarily from the order Glomerales, and relatively little is known of the ecology of other orders including the Paraglomerales. We investigated the distribution of the Paraglomerales across the English agricultural landscape under different management systems. Soils were collected from 11 tilled agricultural sites. Presence of Paraglomerales was assessed using PCR amplification of 18S/ITS region ribosomal DNA isolated from trap plants, terminal restriction fragment length polymorphism and cloning. Paraglomus spp. were detected in all samples from one location and sporadically in six more, but not at the other locations. Distribution was not related to soil physiochemical characteristics, but the Paraglomaceae were significantly more common in soils under organic management. Cloning of samples from three sites produced sequences closely related to Paraglomus laccatum but only distantly related to Paraglomus brasilianum and Paraglomus occultum. Individual sites had between 10 and 27 separate terminal restriction fragments (T-RFs). The large number of T-RFs reflected a significant sequence diversity in the ITS region. Paraglomerales were, therefore, widely distributed across the agricultural landscape, though with patchy distribution and low diversity. More intensive agricultural management appeared to impact negatively on Paraglomus spp.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Arbuscular mycorrhizal (AM) fungi form a symbiotic relationship with around 80 % of plant families and are an important part of the soil microbial community. The host plant may benefit from reduced damage by soil pathogens (Lingua et al. 2002; Pozo et al. 2002), improved water relations (Augé et al. 2004), increased uptake of some micronutrients (Faber et al. 1990; Kothari et al. 1991) and improved soil structure, (Degens et al. 1996), although the main benefit AM fungi confer is an increase in host P uptake (Smith and Read 1997).
Despite the importance of AM fungi, little was known of their ecology until relatively recently because of the inability to identify individual species growing within plant roots. The advent of PCR-based molecular techniques in the last decade has revolutionised the study of AM fungi and led to new insights into their role in structuring plant communities as well as their taxonomy. However, molecular techniques have their limitations, one of which is the lack of a universal primer set which can amplify all AM families while excluding non-AM fungi. Although attempts have been made to develop universal primer sets, they either lack practicality (Kruger et al. 2009) or fail to capture all families (Lee et al. 2008). As a result of their dominance in most ecosystems, primers have tended to focus on the Glomeraceae, meaning that the ecology of other families has been investigated to a lesser degree. One of the earliest and still most commonly used AM-specific primer sets AM1/NS31 fails to amplify DNA from some AM fungal orders including the Paraglomerales (Daniell et al. 2001; Schüßler et al. 2001). Indeed, there has been some difficulty in developing primers specific to this group (Renker et al. 2003; Wirsel 2004). This failing is compounded by the fact that spores of Paraglomus spp. are very similar to those of some Glomus species in the order Glomerales, resulting in traditional spore-based techniques of identifying AM fungi also often failing to identify the presence of Paraglomus species. As a result, Paraglomus-type spores have often been grouped with other spores in the literature (e.g. ‘Glomus occultum… and G. occultum-like spores’, Galvez et al. 2001; ‘G. occultum, Glomus aggregatum-like and Glomus microaggregatum’, Franke-Snyder et al. 2001; ‘comprises Paraglomus occultum and Glomus albidum’, Oehl et al. 2003). Indeed the Paraglomerales were not recognised as a separate group until molecular based phylogenetic techniques became available (Morton and Redeker 2001). Though morphologically similar to some Glomerales and previously classified as Glomus species, they are now recognised as being phylogenetically distant. The order is monogeneric, and currently, there are just four species recognised in a single genus Paraglomus within the family Paraglomeraceae, and these are P. occultum, P. brasilianum, P. laccatum and the recently described P. majewskii (Blaszkowski et al. 2012). They remain a poorly investigated group, and there is some doubt about their diversity, particularly whether all cultures identified as P. occultum are conspecific, and as a result, there may be greater diversity within the Paraglomerales than is currently recognised (Stockinger et al. 2009). While P. brasilianum has distinctive spores, those of the other three species are more similar and less easy to differentiate (Blaszkowski et al. 2012). Recent primer development (Hijri et al. 2006; Lee et al. 2008; Krueger et al. 2009) has enabled Paraglomus spp. to be successfully amplified, but little data have yet to emerge in the literature regarding their ecology, and they remain a neglected group. What evidence we do have suggest that the Paraglomerales may have an interesting ecology, with Paraglomus spp. forming a significant component of the AM community at some sites (Kurle and Pfleger 1996; Oehl et al. 2003) while being absent or rare at others (Hendrix et al. 1995; Kurle and Pfleger 1996; Franke-Snyder et al. 2001; Galvez et al. 2001; Hijri et al. 2006; Lumini et al. 2010; Drumbell et al. 2011). In this work, we sought to address the question of how widely distributed species of Paraglomus are in tilled agricultural soils, which of the four species is most common and the extent to which agricultural management and soil properties influence their distribution.
Methods and materials
Sites
Eleven sites were selected across England (Table 1, Fig. S1) including arable, arable/horticultural and horticultural farms. Arable cropping was mainly small grains, wheat (Triticum spp. L.) and barley (Hordeum vulgare L.) with break crops of either beans (Vicia spp. L.) or oil seed rape (Brassica napus L.). Horticultural cropping consisted predominately of vegetables, including brassicas, with occasional small grain break crops, while horticultural/arable rotations were a mixture of small grains with occasional vegetable crops, usually potatoes (Solanum tuberosum L.). Soil type varied between sites (Table 1). Inversion tillage was the primary form of tillage at all sites.
At each site, a field managed under organic principles and a field managed conventionally, but with a similar cropping history, were selected. The selection of paired organic and conventional fields was intended to address the question of whether different agricultural practices influence the occurrence of Paraglomus spp. in agricultural soils. Soil types were identical in the two fields at each site.
Soil collection and analysis
Soils were sampled in February 2003. In each field, five contiguous 10 × 10-m plots were marked out as a transect. From within each plot, 20 soil cores (0–30 cm) were collected with a 5-cm diameter soil auger and pooled to provide a sample approximately 5 kg in weight. Each soil sample was passed through a 6-mm sieve before analysis for total organic carbon (acid dichromate wet oxidation), total P (acid digestion (aqua rega/HF)), extractable P (Olsen 1954) and pH (H2O) in a soil to solution ratio of 1:5. The soils showed a spectrum of pH, organic matter and total and available P contents (Table 2).
Identification of Paraglomerales
In order to assess whether Paraglomus species were present and active in the soils from each field, a trap plant method was employed. This method was chosen in preference to direct sampling of field roots to standardise AM sampling conditions between sites and fields as much as possible and to remove differences in crop plant and climatic variables as confounding factors (van der Gast et al. 2011). Direct sampling of field soil was rejected because we anticipated that the Paraglomerales would be rare, and therefore, direct extraction of soils involving small samples of less than 1 g might potentially miss propagules or colonised root fragments. Our trap plants in contrast were able to ‘sample’ 200 g of soil.
Soils were adjusted to 40 % water holding capacity (WHC) and 200 g fresh weight placed into plastic plant pots. Three onion seeds (Allium cepa cv. Balstora) were sown into each pot, and individual pots were sealed in Sunbags (Sigma Chemical Co., Poole, Dorset) to prevent cross-contamination (Walker and Vestberg 1994). The pots were placed in a glasshouse in a randomised block design. Glasshouse temperature was maintained under a 20/15 °C day/night regime. Pots were weighed weekly and watered with deionised water to maintain soil at 40 % WHC. Seedlings were harvested after 14 weeks, and the roots and shoots were separated. Roots were washed in deionised water, cut to approximately 2 cm lengths and mixed thoroughly. Approximately 500 mg of roots were retained for DNA extraction. In order to determine if Paraglomus species had colonised trap plants, a combination of terminal restriction fragment length polymorphism (T-RFLP) of 18S/ITS region ribosomal DNA (rDNA) and cloning and sequencing was employed.
DNA was extracted from 100 mg root samples using the DNeasy Plant mini kit (Qiagen, Crawley, UK) following the manufacturer's instructions. 18S/ITS rRNA gene fragments were amplified using the specific primer Para1313, with the universal primer ITS4. Though a number of primers had been published at the time this work was conducted, which purport to amplify AM fungi from the Paraglomerales (e.g. Redecker 2000), we found that some lack specificity, producing many false positives, even with pure cultures, a result supported by other authors (Wirsel 2004). In contrast, we found the primer pair Para1313/ITS4 (Hijri et al. 2006) to be effective in amplifying Paraglomerales. Initial PCR amplification was conducted using the general eukaryotic primers NS5 and ITS4 (White et al. 1990) in a 25-μl reaction mixture containing 0.25 μl of each primer (25 ρm μl−1), 1 μl of bovine serum albumin (BSA), 21 μl of MegaMix (Microzone Ltd.) and 2.5 μl neat DNA, using conditions described in Hijri et al. (2006). Products from this PCR were visualised on a 1.3 % agarose gel. Where a band of the expected size (c. 1,500 bp) was visible, product from the first PCR was diluted 1:100 in water or 1:1,000 where the band was very strong. Where no band was visible, product was diluted 1:10. Diluted product was then used as a template in a nested PCR using the primers ITS4 and Para1313 (Redeker et al. 2003; Hijri et al. 2006), and reaction mixture was as follows: 1 μl each of ITS4 and Para1313 Hex and 6-carboxyfluorescein (FAM) fluorescent labelled, respectively (25 ρm μl−1), 1 μl of BSA, 42 μl of MegaMix and 5 μl of DNA. Reaction programme was follows: 1 cycle for 3 min at 95 °C, 35 cycles of 45 s at 95 °C, 50 s at 62 °C and 90 s at 72 °C, followed by a final extension of 10 min at 72 °C.
Products from the second PCR were visualised on a 1.3 % agarose gel. Where a product of the correct size (c. 1,100 bp) was identified, T-RFLP was conducted as follows: PCR products were cleaned using the QIAquick PCR purification kit (Qiagen, Crawley, UK) and then digested with the restriction endonuclease MboII (Promega, Madison, USA) at 37 °C for 4 h. This restriction enzyme was shown to be the most effective at providing diagnostic T-RF for published sequences of P. occultum (AJ006799) and P. brasilianum (AJ301862) using the Restriction Enzyme Mapping Application software package (http://bioperl.macaulay.ac.uk/startForm.htm), with P. occultum generating a unique T-RF at 143 bp and P. brasilianum at 434 bp, and both strains giving a T-RF at 221 bp. Published sequences across the Para1313 and ITS4 regions were not available to provide comparative information for P. laccatum or P. majewskii.
Digestion products were purified and run on the Applied Biosystems 377 automated DNA sequencer. Data from T-RFLP were analysed with the aid of the programme GeneMarker® v1.5 (SoftGenetics.com) for peak calling. As most T-RFLP traces were ‘clean’ with very low noise levels, there were no size or area cut-offs used for peak calling, and all clear peaks were included in the analysis provided that a peak of that size occurred in at least two samples. The exception was those peaks below 75 bp in size which were excluded because of uncertainty in distinguishing true peaks from the primer peak.
Verification of bioassay and molecular profiling approaches using Paraglomus cultures
In order to be sure that our methodology was able to detect and identify the described species of Paraglomus, we obtained pure cultures of three of the four described Paraglomus species in the form of a mixture of sand, root fragments and spores. For P. brasilianum and P. occultum, cultures BR105 and IA707, respectively, were obtained from the International Collection of Arbuscular Mycorrhizal Fungi at the University of West Virginia, USA. For P. laccatum, culture Attempt-694 was a gift from Dr. Chris Walker.
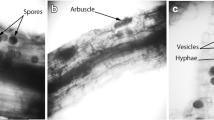
Inoculum was added to a mixture of 50/50 w/w sand and Terragreen in 9-cm plastic pots. Into the inoculated mixture, pre-germinated onion seeds were planted, and the pots were placed in sunbags and treated as described above. After 30 weeks, plants were harvested. Roots were washed in deionised water and examined under a dissecting microscope for evidence of colonisation after staining with aniline blue (Grace and Stribley 1991). In addition, samples of the sand–Terragreen mixture were washed through a series of sieves onto a 38-μm sieve, and the material retained on this sieve was examined for AM fungal spores.
To determine the possibility of primer bias adversely impacting on our results, we sought to determine that our reaction conditions could amplify Paraglomus species when they were present in roots. DNA was extracted from 600 mg samples of onion roots and sand–Terragreen colonised by each of the individual Paraglomus species used to assess colonisation potential. In addition, DNA extractions were carried out on mixtures of roots colonised by three species. Approximately 200 mg of roots and sand–Terragreen of each of the three species were extracted together as described above. PCR reactions were carried out on mixed and single-species DNA extractions as described above. DNA, extractions and PCR and T-RFLP reactions were carried out in triplicate using the procedures described above.
Cloning reactions and sequence analysis
To confirm that our PCR reaction was amplifying only Paraglomus rDNA and to verify results of T-RFLP, PCR products from three fields were cloned. DNA from the Wellesbourne conventional field, the Duggleby conventional field and the Tarlton organic field was amplified as described above with unlabeled primers. Duplicate reactions were completed for each site, in order to obtain sufficient DNA, and pooled before PCR clean-up. Analysis of T-RFs from across the sites indicated that most T-RFs obtained in the study were found at these sites. PCR product was cloned using the Qiagen PCR cloning kit (Qiagen, UK). Sequencing reactions were conducted using the PRISM BigDye Terminator Cycle Sequence reaction kit (Applied Biosystems, Foster City, USA), with products analysed on the Applied Biosystems 377 automated DNA sequencer. DNA sequences were edited and assembled using the DNAstar II sequence analysis package (Lasergene, Inc., Madison, USA). Sequences were compared with those on the European Molecular Biology Laboratory (EMBL) DNA database. T-RF lengths were calculated for cloned sequences using the Restriction Enzyme Mapping Application software package.
Data analysis and statistics
All statistical analysis was carried out in GenStat (GenStat 2007, tenth edition) unless otherwise stated. The number of fields showing PCR bands under conventional and organic management was compared using paired t test. All analyses of T-RFLP bands were done on ITS4 Hex-labelled fragments only, as these showed a much greater diversity, both from in silico digests of clone sequences and in the T-RFLP data. Mean number of peaks per sample was compared with two-way ANOVA (site and management practice), and the relationship between the number of T-RFs per sample and the soil parameters was explored using partial Pearson correlation. Estimates of true T-RF diversity present in each field were made using the EstimateS 8.0.0 (Colwell 2006). Chao 1, Chao 2 and Jackknife 1 and 2 were calculated without replacement using 50 randomisations.
Structure of the T-RF ‘populations’ was explored using the peak presence/absence data. Sorensen (Bray–Curtis zero adjusted) similarity coefficients were calculated between individual fields, and clustering of fields was visualised using non-metric multidimensional scaling (NMDS). NMDS is an unconstrained ordination method which has the advantage of being highly robust to violation of assumptions (Minchin 1987). Significance of clustering in NMDS was assessed using multi-response permutation procedure (MRPP) again using Sorensen similarity. At two sites with multiple positive samples (Duggleby, Ryton and Wellesbourne), differences in a community structure between conventional and organic fields were explored further using NMDS and MRPP as described above, and all calculations were carried out in PC-ORD version 5.06 (McCune and Mefford 2006).
The phylogenetic relationship of clones obtained from Wellesbourne, Duggleby and Tarlton with database sequences was explored using the Phylip software package (version 3.67; Felsenstein 2007). Sequences were aligned in MegAlign 5.03 (DNAStar, Inc., USA) and checked manually. Sequences were bootstrapped to give 1,000 replications and then compared using the Kimura two-parameter model to calculate distances with gamma distribution set at 0.5. The neighbour-joining method was used to construct phylogenetic trees, and then a consensus tree was arrived at. No published sequence data for P. majewskii are currently available for the ITS region.
Results
All three tested species of Paraglomus successfully colonised onion roots in the sand/Terragreen mixture using single-species inoculum. Examination of roots showed evidence of characteristic mycorrhizal structures, hyphae and vesicles. In addition, spores were extracted from P. brasilianum and P. occultum cultures by sieving. They were small and hyaline, characteristic of Paraglomus species, but sporulation of P. laccatum was not evident, and no spores were seen. PCR of roots from the three individual species was successful. Several different T-RFs over 75 bp were seen in T-RFLP profiles of P. brasilianum BR105 (142, 143 and 253 bp) and P. occultum IA707 (238 bp); although no T-RF was seen for P. laccatum strain 695, T-RFLP profiles of the mixed DNA extractions included the T-RF characteristic of both P. brasilianum and P. occultum, indicating that a primer bias should not significantly influence our results.
Of the 109 samples of roots from onions grown on field-collected soil, PCR product of the correct size was evident in 35 (Table 3). Previous work using the NS31/AM1 primer set (Simon et al. 1992; Helgason et al. 1998) has shown that 98 of these 109 samples contain DNA from the Glomales or Diversisporales (van der Gast et al. 2011), with nine samples having DNA neither from the Paraglomerales nor Glomales or Diversisporales. This indicates both the relative rarities of these Paraglomus spp. over the landscape and suggests that factors causing the absence of Glomales and Diversisporales from a site similarly impact on the Paraglomerales, as no sites contained Paraglomerales but not Glomales or Diversisporales.
At sites where DNA from Paraglomus spp. was identified, the frequency of occurrence varied markedly. At the Wellesbourne and Duggleby sites, all plots produced a positive result (except one), while at the Tarlton site, all plots in the organic field produced a positive result, but soil from just one plot in the conventional field produced a positive result. Evidence of Paraglomerales in soils from other sites was sporadic, with no field producing a positive result for these species from all samples. At three sites, there were some positive results from samples from the organic field, but no positive results from the conventional fields. In contrast, at no site did the conventional field produce a positive result, and the organic soil, a negative one. Overall, there was a significantly greater occurrence of a positive product in the organic soils than the conventional (P < 0.03). At three sites (Epworth, Kirton and Great Coxwell), no plants grown on soils from either the organic or conventional field produced a positive result.
T-RFs occurred in all samples where a PCR product of the correct size was identified on agarose gel, although T-RFs with a FAM label (Para1313 end) were absent in many samples including all the Duggleby organic and Ormskirk samples, suggesting that sequences had a restriction site close to the primer. As a result, all subsequent analyses were done on the Hex (ITS4)-labelled fragments, although in the case of one Fosdyke sample and one Ormskirk sample, the only Hex-labelled peaks were less than 75 bp. These samples were not included in further analyses because of the possibility of confusing small T-RFs with the primer peak.
There were a large number of T-RFs identified in many samples. Soils from most fields produced more than 20 different T-RFs, with two sites producing more than 30 (Table 3). Estimates of true richness were generally close to measured richness for each field (Table S1), indicating that our sampling effort was sufficient to capture most of the sequence diversity. There was no significant difference in the mean number of T-RFs between conventional and organic management systems where both organic and conventional fields produced a product (P 0.7). The difference in the mean number of T-RFs between sites was significant (P 0.02).
Soil characteristics did not appear to have an influence on the distribution of Paraglomus spp., despite large differences between sites, particularly for soil organic matter and extractable P (Table 2). There was no common feature linking soils either with or without Paraglomus spp., and there were no significant correlations between the number of T-RFs and any of the soil physicochemical characteristics, including pH, organic matter and total and available P contents.
Examination of the actual T-RFs obtained from different sites and fields revealed no obvious clustering of conventional and organic fields (Fig. 1), and MRPP confirmed that management system had no significant effect on clustering (P 0.3). The effect of conventional versus organic management was explored further at the three sites (Duggleby, Ryton and Wellesbourne), where there were multiple positive results in conventional and organic fields. Duggleby was the only site where organic and conventional T-RF patterns clustered separately (data not shown, MRPP, P 0.04). When the structure of T-RF patterns at different sites was compared (i.e. conventional and organic communities combined), there was found to be a considerable overlap; however, separation between sites was significant (MRPP, P 0.03), indicating that different locations supported different T-RF communities (Fig. 2).

Non-metric multidimensional scaling ordination plot showing relationship between T-RFs in conventional (empty circle) fields with organic (filled circle) fields. In the result of 500 iterations, 250 each are with real and randomised data (Monte Carlo test). Final stress for two dimensions is 20.62; final instability, 0.000. Variation in distance matrix is represented by axes 1 374.4 %, 2 13.9 %

Plot of non-metric multidimensional scaling ordination comparing Paraglomus T-RF patterns at different sites, organic and conventional fields combined for each site
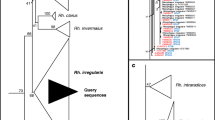
A combined total of 117 clones were successfully sequenced from the Wellesbourne, Duggleby and Tarlton sites. The clones showed a high degree of similarity to Paraglomus sequences contained in the EMBL database, with no evidence of contamination by other mycorrhizal families or non-mycorrhizal fungi. Figure 3 shows the relationship between representative clones from Wellesbourne, Duggleby and Tarlton with named accessions and environmental sequences produced by Hijri et al. (2006) using the same primer set, based on a 450-bp fragment of our cloned sequences from the ITSI/II and 5.8S region. Sequences were deposited in the EMBL database with designated accession numbers from FN555262 to FN555292. Clones from Wellesbourne and Duggleby were most closely related to P. laccatum AM29549 and were also very similar to environmental sequences obtained from an arable field in Switzerland (Hijri et al. 2006), showing more than 94 % similarity in all cases. Cloned sequences from Tarlton formed a separate clade from the described species and sequences from Wellesbourne, Duggleby and Switzerland. This clade was highly supported by the bootstrap analysis, >97 %, but these sequences were also closely related to P. laccatum, showing less than 5 % divergence, compared with at least 15 % divergence from both P. occultum or P. brasilianum sequences.

Phylogenetic tree of Paraglomus showing the relationship of sequences from Wellesbourne (Well), Tarlton (Tarl) and Duggleby (Dug) soils to named species and environmental sequences obtained from arable fields in Switzerland (Hijri et al. 2006), using also the PARA1313/ITS4i primer set. (Note that Paraglomus species were previously classified as Glomus species, and the database sequences included here still appear on databases under the genus Glomus). Tree is based on approximately 450 characters in the ITSI and II and 5.8S regions, bootstrapped with 1,000 replications, using the neighbour-joining method. Distance relationships were obtained using the Kimura two-parameter model, with a gamma distance correction of 0.5 used. Numbers before named sequences refer to accession numbers in EMBL database as do numbers after clone names from the three sites, while numbers in brackets refer to terminal restriction fragment sizes for those sequences (PARA1313 and ITS4 ends, respectively). Numbers above branches refer to bootstrap values
There was a generally good agreement between the size of the largest T-RF peaks and the size of T-RFs obtained from the cloned sequences. Matches were within the normal range encountered with T-RFLP (Burke et al. 2005; Dickie and Fitzjohn 2007). The large number of different T-RFs derived from clones was the result of multiple base insertions/deletions in the ITS regions of the sequences. The 5.8S region was much less variable in our clones.
Table 4 shows the distribution across sites of those T-RFs which corresponded with the size of T-RFs derived from clones from the Wellesbourne, Duggleby and Tarleton sites. Some T-RFs such as of 170 and 194 bp were more common in organic fields, while none appeared to be more associated with conventional fields. T-RFs of 401, 404 and 405 bp were ubiquitous, occurring in all fields where Paraglomerales were detected, while peak 196 only occurred at two sites.
Discussion
This work demonstrates that Paraglomerales are geographically widespread in tilled agricultural soils in England, occurring over a wide range of soil types and production systems, including organic, conventional, arable and horticultural, but that they may be absent from some locations. However, neither soil P, pH nor soil organic matter appeared to influence the occurrence of Paraglomerales across sites. Although other authors have connected the occurrence of Paraglomus species to soil factors, notably soil P (Kurle and Pfledger 1996; Hijri et al. 2006); their conclusions are contradictory, and our results, using multiple sites, suggest no relationship between soil characteristics and the presence of one or more Paraglomus species. However, our results do indicate that management system has an influence, with Paraglomerales being more common in organically managed soils than conventionally managed soils. Douds et al. (1995) also found the Paraglomerales to be more common under organic management, based on results from one site, although other authors have found no systematic difference (Oehl et al. 2003; Hijri et al. 2006). The absence of any field here where Paraglomerales were detected, but neither Glomerales nor Diversisporales were detected (van der Gast et al. 2011), indicates that the Paraglomerales response to agricultural management is broadly in line with other groups of AM fungi which generally show a negative response to agricultural management (Helgasson et al. 1998), with conventional management being more deleterious than organic (van der Gast et al. 2011). Factors driving the difference between organic and conventional management systems are likely to be multiple but may include rotation, soil fertility management, tillage and crop protection technologies (Gosling et al. 2006).
The lack of difference in the mean number of T-RFs between the conventional and organic management systems at sites where they occurred in both fields, and the lack of difference in the actual T-RFs present was surprising, given the apparent negative impact of conventional production on the occurrence of Paraglomus spp. The exception was the Duggleby site, where there was a clear differentiation between the T-RFs found in each field. This could suggest that different species of Paraglomus at each site were responding in the same way to management practices.
Although the very large number of different T-RFs is indicative of high sequence diversity, the clone data suggest that this diversity, though real, is likely to reflect variability within individual species of Paraglomus and, in the case of the three cloned sites, P. laccatum. There is good evidence for significant rDNA sequence diversity within ITSI and II regions of the Glomeromycota (Stockinger et al. 2009). Sanders et al. (1995), for example, identified two different ITS sequences within an individual spore of Glomus mosseae, while Croll et al. (2008) identified multiple ITS sequences in different individuals and populations of Glomus intraradices. Within the Paraglomerales specifically, Nilsson et al. (2008) recorded 3.2 % diversity within a 550-bp region covering ITSI and II and 5.8S regions in six sequences of P. brasilianum and 19.4 % in 12 sequences of P. occultum, although Stockinger et al. (2009) questioned whether the P. occultum cultures were conspecific. Given this high sequence diversity, it is difficult to be certain whether or not any of the other sites contain species other than P. laccatum or more than one species of Paraglomus. There is a need for greater understanding on intraspecific diversity in Paraglomus spp. which requires single-spore studies across reference isolates.
We are not aware of any instances reported in the literature where more than one species of Paraglomus have been recorded at a single site under agricultural management, including high-throughput sequencing studies (Lumini et al. 2010), although difficulties in correctly identifying Paraglomus spp. from spore morphology leave a question mark over spore-based studies. Our clone data at three sites, together with T-RFLP data from the other eight sites at which Paraglomus spp. was detected, suggest that P. laccatum was the dominant Paraglomus sp. in tilled agricultural soil. Furthermore, studies in a German agricultural soil (Hijri et al. 2006) and an Italian pasture soil (Lumini et al. 2010) found that Paraglomus was represented exclusively by P. laccatum. The fact that spore-based work reports almost invariably the presence of P. occultum is indicative of the limitations of spore-based studies for this particular family of AM fungi.
Trap plants are known to be colonised by different AM fungal communities than field-grown plants (Hazard et al. 2013), and this could have resulted in underestimation of the diversity of the Paraglomus spp. communities. Clearly, the use of high-throughput sequencing approaches (e.g. Lumini et al. 2010) to provide detailed resolution of the structure of root and soil fungal communities provides opportunities to resolve the distribution and diversity of rarer AM fungal taxa such as Paraglomus. If we are only dealing with a single species at each site, then the diversity evident in the T-RFLP data may not be ecologically meaningful. However, at the Duggleby site, different T-RFs were distributed unevenly between the conventional and organic fields, suggesting some niche differentiation. Munkvold et al. (2004), Koch et al. (2006) and Croll et al. (2008) have all demonstrated significant differences in the degree of host response to different isolates of the same morphospecies of several AM fungi, indicating functional diversity within species, thus suggesting that at least some of our T-RF diversity may represent functionally important differences.
Another interesting aspect of our results was the separation of the clones from the Tarlton site into a different clade from those from the Wellesbourne and Duggleby sites and from an arable field in Switzerland. The relatively high degree of similarity in the context of high rDNA sequence diversity within ITSI and II regions indicates a high degree of relatedness, and thus, that all were P. laccatum, but there was a high bootstrap support for the two clades. Host range was similar at these sites (arable cropping), but the soil at Tarlton was a mildly acidic peat, quite distinct from the mineral soils at Wellesbourne, Duggleby and at the site in Switzerland. Whether this was influential on the development of a different lineage is unclear, but it may suggest, as with the differences in T-RFLP patterns between fields at Duggleby, some degree of niche separation between different lineages of the same species.
In conclusion, Paraglomus spp. were shown to be geographically widespread though locally sporadic in tilled agricultural soils. Major soil nutrient concentrations, organic matter and pH were not indicative of the likelihood of the occurrence of Paraglomus spp. or of the occurrence of specific T-RFs and by inference sequences, but relative to conventional management, soils managed organically were more likely to contain Paraglomus spp. T-RF patterns showed spatial structure, with a tendency to differ between sites, suggestive of a degree of niche differentiation, but specific groups of T-RFs were not associated with production systems. A large number of different T-RFs were present at each site, with a good match to cloned sequences, indicative of a high degree of sequence variability. However, only a single species of Paraglomus was present in clone libraries.
References
Augé RM (2004) Arbuscular mycorrhizae and soil/plant water relations. Can J Soil Sci 84:373–381
Blaszkowskii J, Kovacs G, Gaspar BK, Balazs TK, Buscot F, Ryszka P (2012) The arbuscular mycorrhizal Paraglomus majewskii sp.nov represents a distinct basal lineage in Glomeromycota. Mycologia 104:148–156
Burke DJ, Martin KJ, Rygiewicz PT, Topa MA (2005) Ectomycorrhizal fungi identification in single and pooled root samples: terminal restriction fragment length polymorphism (TRFLP) and morphotyping compared. Soil Biol Biochem 37:1683–1694
Cowel RK (2006) EstimateS: statistical estimation of species richness and shared species from samples, version 8.0
Croll D, Wille L, Gamper HA, Mathimaran N, Lammers PJ, Corradi N, Sanders IR (2008) Genetic diversity and host plant preferences revealed by simple sequence repeat and mitochondrial markers in a population of the arbuscular mycorrhizal fungus Glomus intraradices. New Phytol 178:672–687
Daniell TJ, Husband R, Fitter AH, Young JPW (2001) Molecular diversity of arbuscular mycorrhizal fungi colonising arable crops. FEMS Microbiol Ecol 36:203–209
Degens BP, Sparling GP, Abbott LK (1996) Increasing the length of hyphae in a sandy soil increases the amount of water-stable aggregates. Appl Soil Ecol 3:149–159
Dickie IA, FitzJohn RG (2007) Using terminal restriction fragment length polymorphism (T-RFLP) to identify mycorrhizal fungi: a methods review. Mycorrhiza 17:259–270
Douds DD, Galvez L, Janke RR, Wagoner P (1995) Effect of tillage and farming system upon populations and distribution of vesicular-arbuscular mycorrhizal fungi. Agricult Ecosyst Environ 52:111–118
Dumbrell AJ, Ashton PD, Aziz N, Feng G, Nelson M, Dytham C, Fitter AH, Helgason T (2011) Distinct seasonal assemblages of arbuscular mycorrhizal fungi revealed by massively parallel pyrosequencing. New Phytol 190:794–804
Faber BA, Zasoski RJ, Burau RG, Uriu K (1990) Zinc uptake by corn as affected by vesicular-arbuscular mycorrhizae. Plant Soil 129:121–130
Felsenstein J (2007) PHYLIP (Phylogeny Inference Package) version 3.67. Distributed by the author. Department of Genetics, University of Washington, Seattle
Franke-Snyder M, Douds DD, Galvez L, Phillips JG, Wagoner P, Drinkwater L, Morton JB (2001) Diversity of communities of arbuscular mycorrhizal (AM) fungi present in conventional versus low-input agricultural sites in eastern Pennsylvania, USA. Appl Soil Ecol 16:35–48
Galvez L, Douds DD, Drinkwater LE, Wagoner P (2001) Effect of tillage and farming system upon VAM fungus populations and mycorrhizas and nutrient uptake of maize. Plant Soil 118:299–308
GenStat (2007) Genstat for Windows, release 10.1.0.71, tenth edition. VSN International Ltd., Oxford
Gosling P, Hodge A, Goodlass G, Bending GD (2006) Arbuscular mycorrhizal fungi and organic farming. Agricult Ecosyst Environ 113:17–35
Grace C, Stribley DP (1991) A safer procedure for routine staining of vesicular-arbuscular mycorrhizal fungi. Mycol Res 95:1160–1162
Hazard C, Gosling P, Mitchell DT, Doohan FM, Bending GD (2013) Landscape-scale distribution of arbuscular mycorrhizal fungal communities is affected by the local environment, but not geographical distance. ISME J 7:498–508
Helgason T, Daniell TJ, Husband R, Fitter AH, Young JPW (1998) Ploughing up the wood-wide web? Nature 394:431
Hendrix JW, Guo BZ, An Q (1995) Divergence of mycorrhizal fungal communities in crop production systems. Plant Soil 170:131–140
Hijri I, Sykorova Z, Oehl F, Ineichen K, Mader P, Wiemken A, Redecker D (2006) Communities of arbuscular mycorrhizal fungi in arable soils are not necessarily low in diversity. Mol Ecol 15:2277–2289
Koch AM, Croll D, Sanders IR (2006) Genetic variability in a population of arbuscular mycorrhizal fungi causes variation in plant growth. Ecol Lett 9:103–110
Kothari SK, Marschner H, Römheld V (1991) Effect of a vesicular arbuscular mycorrhizal fungus and rhizosphere micro-organisms on manganese reduction in the rhizosphere and manganese concentrations in maize (Zea mays L.). New Phytol 117:649–655
Krüger M, Stockinger H, Krüger C, Schüßler A (2009) DNA-based species level detection of Glomeromycota: one PCR primer set for all arbuscular mycorrhizal fungi. New Phytol 183:212–223
Kurle JE, Pfleger FL (1996) Management influences on arbuscular mycorrhizal fungal species composition in a corn-soybean rotation. Agron J 88:155–161
Lee J, Lee S, Young JPW (2008) Improved PCR primers for the detection and identification of arbuscular mycorrhizal fungi. FEMS Microb Ecol 65:339–349
Lingua G, D'Agostino G, Massa N, Antosiano M, Berta G (2002) Mycorrhiza-induced differential response to a yellows disease in tomato. Mycorrhiza 12:191–198
Lumini E, Orgiazzi A, Borriello R, Bonfante P, Bianciotto V (2010) Disclosing arbuscular mycorrhizal fungal biodiversity in soil through a land-use gradient using a pyrosequencing approach. Environ Microbiol 12:2165–2179
McCune B, Mefford MJ (2006) PC-ORD. Multivariate analysis of ecological data, version 5.06. MjM Software, Gleneden Beach, Oregon
Minchin PR (1987) An evaluation of relative robustness of techniques for ecological ordinations. Vegetatio 69:89–107
Morton JB, Redecker D (2001) Two new families of Glomales, Archaeosporaceae and Paraglomaceae, with two new genera Archaeospora and Paraglomus, based on concordant molecular and morphological characters. Mycologia 93:181–195
Munkvold L, Kjoller R, Vestberg M, Rosendahl S, Jakobsen I (2004) High functional diversity within species of arbuscular mycorrhizal fungi. New Phytol 164:357–364
Nilsson RH, Kristiansson E, Ryberg M, Hallenberg N, Larsson KH (2008) Intraspecific ITS variability in the kingdom fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol Bioinform 4:193–201
Oehl F, Sieverding E, Ineichen K, Mader P, Boller T, Wiemken A (2003) Impact of land use intensity on the species diversity of arbuscular mycorrhizal fungi in agroecosystems of Central Europe. Appl Environ Microbiol 69:2816–2824
Olsen SR, Cole CV, Watanabe FS, Dean LA (1954) Estimation of available phosphorus in soils by extraction with sodium carbonate. US Department of Agriculture, Circular no, 939
Pozo MJ, Cordier C, Dumas-Gaudot E, Gianinazzi S, Barea JM, Azcon-Aguilar C (2002) Localized versus systemic effect of arbuscular mycorrhizal fungi on defence responses to Phytophthora infection in tomato plants. J Exp Bot 53:525–534
Redecker D (2000) Specific PCR primers to identify arbuscular mycorrhizal fungi within colonised roots. Mycorrhiza 10:73–80
Redecker D, Hijri I, Wiemken A (2003) Molecular identification of arbuscular mycorrhizal fungi in roots: perspectives and problems. Filoa Geobotanica 38:113–124
Renker C, Heinrichs J, Kaldorf M, Buscot F (2003) Combining nested PCR and restriction digest of the internal transcribed spacer region to characterize arbuscular mycorrhizal fungi on roots from the field. Mycorrhiza 13:191–198
Sanders IR, Alt M, Groppe K, Boller T, Wiemken A (1995) Identification of ribosomal DNA polymorphisms among and within spores of the Glomales—application to studies on the genetic diversity of arbuscular mycorrhizal fungal communities. New Phytol 130:419–427
Schüßler A, Gehrig H, Schwarzott D, Walker C (2001) Analysis or partial Glomales SSU rRNA gene sequences: implications for primer design and phylogeny. Mycol Res 10:5–15
Simon L, Lalonde M, Bruns TD (1992) Specific amplification of 18S fungal ribosomal genes from vesicular-arbuscular endomycorrhizal fungi colonizing roots. Appl Environ Microbiol 59:291–295
Smith SE, Read DJ (1997) Mycorrhizal symbiosis, 2nd edn. Academic, London
Stockinger H, Walker C, Schüßler A (2009) Glomus intraradices DAOM197198′, a model fungus in arbuscular mycorrhiza research, is not Glomus intraradices. New Phytol 183:1176–1187
van der Gast CJ, Gosling P, Tiwari B, Bending GD (2011) Spatial scaling of arbuscular mycorrhizal fungal diversity is affected by farming practice. Environ Microbiol 13:241–249
Walker C, Vestberg M (1994) A simple and inexpensive method for producing and maintaining closed pot cultures of arbuscular mycorrhizal fungi. Agricult Sci Finland 3:233–240
White TJ, Burns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic, San Diego, pp 315–322
Wirsel SGR (2004) Homogeneous stands of a wetland grass harbour diverse consortia of arbuscular mycorrhizal fungi. FEMS Microbiol Ecol 48:129–138
Acknowledgments
This work was supported by the Department of the Environment Food and Rural Affairs. Thanks also go to those farmers and research institutions that allowed us access to take samples and provided us with management information. Special thanks go to Dr. Chris Walker who supplied single-species inoculum for testing primers and helped with the spore identification and assessment of colonisation in our Paraglomus test plants.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gosling, P., Proctor, M., Jones, J. et al. Distribution and diversity of Paraglomus spp. in tilled agricultural soils. Mycorrhiza 24, 1–11 (2014). https://doi.org/10.1007/s00572-013-0505-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00572-013-0505-z