
Abstract
Purpose
This trial was conducted to confirm the non-inferiority of remimazolam versus propofol in the induction and maintenance of general anesthesia in surgical patients.
Methods
Surgical patients (n = 375) were randomized to remimazolam started at 6 or 12 mg/kg/h by continuous intravenous (IV) infusion until the loss of consciousness (LoC), followed by 1 mg/kg/h to be adjusted as appropriate until the end of surgery or IV propofol administered as a slow bolus of 2.0–2.5 mg/kg until LoC followed by 4–10 mg/kg/h until the end of surgery. Efficacy was measured via the combined primary endpoint of no intraoperative awakening/recall, no need for rescue sedatives, and no body movements. Adverse events and adverse drug reactions (ADRs) were monitored for safety.
Results
Efficacy rates were 100% in all treatment groups, and the non-inferiority of remimazolam was demonstrated [95% confidence interval (− 0.0487; 0.0250)]. The time to LoC was longer in the remimazolam 6 (p < 0.0001) and 12 mg/kg/h (p = 0.0149) groups versus propofol. The time to extubation was longer in both remimazolam groups versus the propofol group (p ≤ 0.0001). The incidence of ADRs was similar in the remimazolam groups (39.3% and 42.7%, respectively) compared with the propofol group (61.3%). Decreased blood pressure occurred in 20.0% and 24.0% of patients treated with 6 and 12 mg/kg/h remimazolam, respectively, compared with 49.3% of patients receiving propofol. Injection site pain was reported in 18.7% of propofol patients but not in those receiving remimazolam.
Conclusions
This trial demonstrated that remimazolam was well tolerated and non-inferior to propofol with regard to efficacy as a sedative hypnotics for general anesthesia.
Clinical trial registration
This trial is registered with the Japan Pharmaceutical Information Center - Clinical Trials Information (JapicCTI). JapicCTI number: 121973
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Propofol and midazolam are the most frequently used intravenous (IV) anesthetics for the induction and maintenance of sedation during general anesthesia [1]. Propofol is often associated with pain on injection [2] and hemodynamic depression; [3] midazolam has a slower onset of action, shows accumulation throughout general anesthesia with prolonged recovery times, is metabolized to an active metabolite, and interacts with all drugs that are metabolized via the cytochrome P450 pathway [4]. Hence, there is an ongoing, unmet need for shorter-acting anesthetics with rapid onset, good control of the depth of anesthesia, full, rapid, and predictable recovery, a benign safety profile particularly in terms of hemodynamic effects, and which are metabolized independently from renal or liver function.
Remimazolam, a full agonist at the benzodiazepine binding site of the gamma-aminobutyric acid (GABA)A receptor, [5, 6] is metabolized by esterases and has a stable context-sensitive half-time of 6–7 min during its administration for various durations [7,8,9,10,11]. Pharmacokinetic modeling showed that remimazolam has a high clearance and small volume of distribution [7,8,9,10,11]. A full clinical development program for procedural sedation has been successfully conducted, [12,13,14,15,16] and Phase II clinical trials conducted on anesthesia for general surgery in Japanese patients and cardiac surgery in European patients indicated that remimazolam is capable of inducing and maintaining sedation during general anesthesia with improved hemodynamic stability [17, 18].
The rationale for this Phase IIb/III, open-label clinical trial was to confirm these capabilities of remimazolam in a broader patient population to obtain approval for a planned indication of induction and maintenance for general anesthesia in Japan.
The key objectives were to evaluate the non-inferior efficacy and safety of remimazolam compared with propofol in surgical patients undergoing general anesthesia using a multicenter, randomized, parallel-group design.
It was tested the hypothesis that remimazolam was non-inferior compared with propofol in terms of functional capacity as a sedative for general anesthesia. The corresponding primary efficacy endpoint was defined as a combination of the following three parameters: (i) intraoperative awakening or recall, (ii) requirement of rescue sedation with other sedatives, and (iii) body movement, all of which are frequently used to clinically assess adequate anesthesia.
Methods
Trial design and patient population
This trial was approved by the Institutional Review Board of Hamamatsu University in Japan, and written informed consent was obtained from all patients prior to participation. The trial was registered before enrollment at the Japan Pharmaceutical Information Center (JapicCTI number: 121973; Principal Investigator: Prof. Shigehito Sato; Date of registration: 26 Sep 2012).
This was a multicenter, single-blind, randomized, parallel-group, Phase IIb/III trial conducted at 49 Japanese sites between November 2012 and March 2013 (Online Resource 1). The trial adhered to the International Conference on Harmonization Good Clinical Practice Guidelines [19] and was conducted in accordance with the Declaration of Helsinki [20].
Relevant inclusion criteria included ≥ 20 years old, a body weight of 100 kg or less, scheduled for elective surgery requiring tracheal intubation and hospitalization for ≥ 3 days (including the days before and after surgery), and an American Society of Anesthesiologists physical status (ASA-PS) of I or II [21]. Key exclusion criteria included emergency surgeries or surgeries expected to last less than 1 h, planned use of extracorporeal circulation, the administration of spinal, epidural, or regional anesthesia between entry into the operating room until extubation, patients undergoing hepatectomy or liver transplant, uncontrolled hypertension (e.g., systolic blood pressure ≥ 160 mmHg on antihypertensive medication), renal impairment (serum creatinine ≥ 2 mg/dL), or hepatic impairment [aspartate aminotransferase (ASAT)/alanine aminotransferase (ALAT) ≥ 2.5 × ULN].
Patients underwent single-blinded randomization (patient-only) to either remimazolam or propofol treatment groups, while the allocation to either of the two remimazolam arms (the 6 mg/kg/h group and the 12 mg/kg/h group) was conducted in a double-blinded (patient plus investigator) manner. For this, the investigator contacted a dedicated patient registration center for eligible patients and requested randomization to one of the three treatment groups (remimazolam 6 mg/kg/h, remimazolam 12 mg/kg/h, or propofol) applying a dynamic allocation with a minimization method and using the following factors: (i) age (20 to < 65 years vs. ≥ 65 years), (ii) ASA-PS (I vs. II), (iii) any planned use of local anesthesia during surgery (Yes vs. No), and (iv) investigational site. This approach was used for an even distribution of patients and to minimize possible biases in the selection of patients and important intergroup imbalances. Masking the anesthesiologist was ensured via two different concentrations in the syringes that were used for induction only, such that the concentration in a syringe for a patient randomized to the remimazolam 12 mg/kg/h dose group was twice as high as that for a patient randomized to the 6 mg/kg/h dose group. After successful induction, the remimazolam syringe was replaced with ones to be used for maintenance, which contained remimazolam at a concentration of 1 mg/mL regardless of the initial randomization result for remimazolam.
Procedures
Induction of general anesthesia
General anesthesia was induced with the investigational product in combination with remifentanil. Muscular paralysis was achieved by rocuronium.
Prior to the induction of anesthesia, oxygen was given for several minutes by lightly placing a mask over the subject’s face. At the same time, remifentanil infusion was started at a rate between 0.25 and 0.5 µg/kg/min and maintained in this range until after intubation.
In the remimazolam groups, the induction of general anesthesia was performed via a continuous infusion of 6 or 12 mg/kg/h for up to 2.5 min. In the propofol group, a bolus of 2.0–2.5 mg/kg was administered slowly over the course of 1 min.
If loss of consciousness (LoC) did not occur after 2.5 min, then the infusion was discontinued, and another sedative was used. LoC was defined as the time when the patient became unresponsive to the shaking of their shoulder.
After LoC was confirmed, an IV dose of rocuronium (0.6–0.9 mg/kg) was administered, and the patient was intubated per hospital protocol.
Maintenance of general anesthesia
During the maintenance phase of anesthesia, remimazolam was administered at a dose of 1 mg/kg/h and adjusted as appropriate (maximum allowed infusion rate: 2 mg/kg/h) based on monitoring of the general condition of individual subjects until the end of the surgery. Propofol was administered at 4–10 mg/kg/h and adjusted as appropriate based on monitoring of the general condition of individual subjects until the end of the surgery.
Remifentanil was continued at an infusion rate between 0.25 µg/kg/min and 2 µg/kg/min and adjusted as considered appropriate. If needed, an additional single intravenous dose of 0.5–1.0 µg/kg remifentanil was allowed.
Rocuronium was administered as necessary as a repeated bolus dose of 0.1–0.2 mg/kg or by continuous intravenous infusion at a dose of 7 µg/kg/min (adjusted as appropriate). Toward the end of the surgery, sugammadex was administered as necessary at a dose of 2–4 mg/kg.
If awakening was not yet observed after 30 min from the end of remimazolam administration, then 0.2 mg flumazenil was given. If necessary, a repeated dose of 0.1 mg was allowed.
The investigators adjusted the investigational products, concomitant drugs, and fluid administrations to maintain a systolic blood pressure ≥ 80 and < 150 mmHg and heart rate ≥ 50 and < 100 bpm. The patients’ depth of sedation was measured by the Bispectral Index (BIS).[22]
If signs of intraoperative awakening (e.g., change in blood pressure or heart rate, lacrimation, or sweating) were noted and assessed as requiring urgent action, then the following procedures were performed:
-
1.
Remimazolam groups: Rapid intravenous infusion of remimazolam at an infusion rate up to 12 mg/kg/h for up to 1 min. If signs of awakening persisted, then remimazolam infusion was discontinued and replaced by another sedative agent.
-
2.
Propofol group: If signs of awakening did not disappear and persisted despite the adjustment of the infusion rate of propofol, then propofol infusion was discontinued and replaced by another sedative agent.
Recovery from general anesthesia
The administration of the investigational product and remifentanil was discontinued at the end of surgery.
The times to eye opening, extubation, stating the date of birth, and decision to exit the operating room were measured and recorded. If eye opening was not observed even after 30 min from the end of remimazolam, then flumazenil was administered.
Surgery was performed according to clinical routine. Participation in the trial did not influence the surgical routine.
Outcomes
Primary endpoint
The primary efficacy endpoint in a given patient was reported for each treatment group and defined as the absence of (1) intraoperative awakening or recall, (2) the need for rescue sedative medication, and (3) body movement. In addition to the patients’ BIS, intraoperative signs of awakening, such as a change in the patients’ blood pressure or heart rate, lacrimation, or sweating, were monitored. Intraoperative recall was assessed using the Brice Questionnaire, [23] which was administered 24 h after surgery and before the patient left the recovery room. Body movement was assessed from LoC until the end of surgery by monitoring the subject for voluntary or purposeful body movement to distinguish these movements from involuntary movement or bucking.
Secondary endpoints
Secondary endpoints included the time to LoC (defined as the time when the subject became unresponsive to the shaking of his/her shoulder) after the start of the investigational medicinal product (IMP).
After stopping the IMP, the time to eye opening, time to accurately state his/her date of birth, time to extubation [defined as the time at which all of the following criteria were achieved: (1) respond to verbal stimuli, (2) adequate respiratory function recovery, (3) stable blood pressure (BP) and heart rate (HR), and (4) recovery of muscle strength (5-s head lift and strong hand grip)], and time to exit the operating room were measured.
The investigator or sub-investigator assessed the controllability of anesthetic depth during the administration of the investigational product using a 3-point scale (1 = Excellent, 2 = Good, and 3 = Poor) shortly after the end of general anesthesia.
Safety parameters
General safety assessments included physical examinations, laboratory testing (hematology, biochemistry, and urinalysis), and vital signs. Non-invasive BP and HR were recorded once during the screening period and immediately before the induction of anesthesia, in 5-min intervals until the patient was transferred out of the operating theatre, and in regular intervals with decreasing frequency until 24 h after surgery. Percutaneous arterial oxygen saturation (SpO2) recordings were started immediately before the induction of anesthesia and continued in 5-min intervals until the patient was transferred to the peripheral ward. A 3- or 5-lead ECG was applied before the induction of anesthesia until recovery and continuously monitored for abnormal clinically relevant changes and relationship to the trial drug. The patients’ agitation level was noted using the Ramsay Sedation Scale Score.
Adverse events (AEs) (defined as any untoward medical occurrence in a patient or worsening of a pre-existing medical condition irrespective of a causal association with the investigational medicinal product) and adverse drug reactions (ADRs) (defined as AEs with a causal relationship to the investigational medicinal product) were monitored throughout this study. Two consecutive recordings of systolic BP either < 80 or ≥ 150 mmHg and HR < 50 or ≥ 100 bpm were recorded as predefined AEs. All AEs were followed until resolution or up to 30 days after the initial follow-up. Laboratory results were compared over time to detect any safety signals.
Statistical analysis
The primary efficacy analysis was based on the Full Analysis Set (FAS), and the safety analysis was based on the Safety Set (SAF) (Online Resource 2). For the composite primary efficacy endpoint, efficacy rates were calculated for all treatment groups, and the between-group differences in rates were calculated with two-sided 97.5% confidence intervals (CIs) using the Wilson method. Non-inferiority was defined as having a CI with a lower limit of more than − 10%. Statistical comparisons were performed using Chi-square tests. In the event that any efficacy rate was 100%, the Newcombe–Wilson hybrid score without continuity correction was used to estimate the 95% CI for the difference between groups.
For secondary endpoints, continuous data were reported as summary statistics and analyzed using t-tests, whereas categorical and ordinal data were reported as frequency distributions and analyzed using Chi-square tests. Time-to-event endpoints were also evaluated using Kaplan–Meier analysis. In the case of AEs, the relative risk and 95% confidence interval were calculated.
Although the use of a single agent for sedation is preferable in the clinical setting, concomitant use of another sedative agent (e.g., additional boluses of propofol during the administration of volatile anesthetics) is sometimes required. Therefore, assuming an efficacy rate of 96% in each group and a non-inferiority margin of 10% for the absolute difference between the groups, the sample size was calculated to observe a greater than − 10% lower limit for the 95% CI of the between-group difference between each of the remimazolam groups and the propofol group (one-sided significance level of 2.5%, 90% power). The statistical analysis of the results was based on a total pool of 375 patients (150 patients for each remimazolam group and 75 patients for the propofol group).
Results
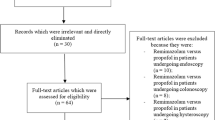
A total number of 391 patients were randomized into 3 groups: 158 to the remimazolam 6 mg/kg/h group, 156 to the remimazolam 12 mg/kg/h group, and 77 to the propofol group. For various reasons (patient decision, investigator decision, or exclusion criteria), 8 subjects in the remimazolam 6 mg/kg/h arm, 6 subjects in the remimazolam 12 mg/kg/h arm, and 2 subjects in the propofol arm did not receive trial medication. Thus, 375 patients were included in these analyses (Fig. 1).

FAS Full Analysis Set, PI Principal Investigator
Overall, baseline demographics and clinical characteristics were comparable. The mean age was 56 years, and the proportion of patients aged 65 or above was 38.7% in the remimazolam 6 mg/kg/h group, 38.0% in the remimazolam 12 mg/kg/h group, and 37.7% in the propofol group. Males accounted for 53% of the patients. Most patients had comorbidities, including vascular, metabolic, respiratory, and nervous system disorders. The most common surgical sites were the limbs and the lower abdomen (Table 1).
The mean duration of surgery was > 2 h in all treatment groups and slightly longer in the remimazolam 6 mg/kg/h group (155.5 min; p = 0.006 vs. propofol) but comparable in the 12 mg/kg/h (143.7 min; p = 0.065 vs. propofol) and propofol (123.4 min; Table 2) groups. Similarly, the mean duration of exposure to remimazolam was longer in the remimazolam 6 mg/kg/h and similar in the 12 mg/kg/h group (202 and 190 min) compared with the propofol group (165 min) (p = 0.003; p = 0.039) (Table 2 and Online Resource 3).
For the composite primary efficacy endpoint, the efficacy rate was 100% in all treatment groups (Table 3). Therefore, the 95% CI was estimated using the Newcombe–Wilson hybrid score. There were no occurrences of intraoperative arousal or recall, need for rescue sedative medication, or body movements in any patients. During the maintenance of anesthesia, BIS values (mean) in the remimazolam 6 mg/kg/h group, remimazolam 12 mg/kg/h group, and propofol group were within the ranges of 40.0–82.0, 47.8–84.0, and 39.0–56.3, respectively (Fig. 2). With a 100% efficacy rate and an estimated 95% CI of [− 0.0487; 0.0250] in all treatment groups, it was therefore assumed that both remimazolam arms were non-inferior to propofol in terms of functional capacity as a sedative for general anesthesia (Table 3).

BIS bispectral index, IMP investigational medicinal product, LoC loss of consciousness
In the remimazolam 6 mg/kg/h group (n = 150) and 12 mg/kg/h group (n = 150), the mean (SD) time to LoC was 102.0 (± 26.6) and 88.7 (± 22.7) seconds, respectively. In the propofol group (n = 75), the mean time to LoC was 78.7 (± 38.4) seconds, indicating a statistically shorter time compared with the remimazolam 6 mg/kg/h and 12 mg/kg/h groups (p < 0.0001 and p = 0.0149, respectively) (Table 3).
During induction, all patients who received remimazolam 6 mg/kg/h reached LoC with cumulative doses lower than 0.3 mg/kg. In contrast, patients who received remimazolam 12 mg/kg/h reached LoC under a broader range of cumulative doses (0.1 to < 1.0 mg/kg). The mean (SD) cumulative dose of remimazolam required to reach LoC was 0.17 (0.04) mg/kg in the 6 mg/kg/h group and 0.29 (0.08) mg/kg in the 12 mg/kg/h arm.
During maintenance, the optimal infusion rate was between 0.8 and 1.0 mg/kg/h for 52% and 58% of patients in the remimazolam 6 and 12 mg/kg/h treatment arms, respectively. For propofol, the optimal rate was between 4.0 and 6.0 mg/kg/h for 52% of patients. Detailed statistics for the induction doses and optimal infusion rates during maintenance are provided in Table 2 and Online Resource 4.
The mean times to eye opening, extubation, stating the date of birth, and decision to leave the operating room were statistically shorter in the propofol group compared with both 6 mg/kg/h and 12 mg/kg/h remimazolam groups (p < 0.05 for all comparisons) (Table 3).
Flumazenil was administered to 9.3% and 8.7% of patients in the remimazolam 6 and 12 mg/kg/h arms, in which the mean time to awakening was 1.8 and 0.9 min after flumazenil administration, respectively.
At least 80% of patients in each treatment group experienced AEs. The most frequently recorded AEs included decreased BP, injection site pain (for 18.7% of propofol patients only), nausea, vomiting, and pyrexia (Online Resource 5). Two patients in the remimazolam 6 mg/kg/h group experienced serious AEs (post-procedural hemorrhage). Neither of these two were regarded as treatment related, and both events were managed with appropriate intervention. No patients discontinued due to AEs, and there were no deaths during the trial.
In contrast, 39.3% and 42.7% of remimazolam 6 mg/kg/h and 12 mg/kg/h patients experienced ADRs compared with 61.3% of propofol patients [RR and 97.5% CI 0.64 (0.47, 0.87) and 0.70 (0.52, 0.94), respectively].
The distributions of AEs and ADRs pertaining to hypotension-specific events were evaluated in a post hoc analysis and are depicted in Fig. 3 and Online Resource 6. For the period up to the completion of intubation, fewer patients in the remimazolam 6 and 12 mg/kg/h groups versus the propofol group experienced ADRs [patients with ADRs 4.7%, 5.3%, and 18.7% (− 14.0 (− 24.5, − 5.4) and − 13.3 (− 23.9, − 4.7)), respectively]. The incidence of ADRs increased after the completion of intubation but remained lower in both remimazolam groups (18.0%, 21.3%) versus 38.7% for propofol [− 20.7 (− 33.2, − 8.3) and − 17.3 (− 30.0, − 4.8)].

***p ≤ 0.0001; **p ≤ 0.0007; *p ≤ 0.009 for remimazolam 12 mg/kg/h vs propofol or remimazolam 6 mg/kg/h vs propofol. Chi-square tests on the between-group difference and 95% Cis (Wilson method) in AE and ADR rates. ADRs adverse drug reactions, AEs adverse events, CI confidence interval, SAF safety set
Systolic and diastolic BP values were higher in the remimazolam groups compared with the propofol group throughout this study period (Online Resource 7). Smaller percentages of patients who received remimazolam 6 and 12 mg/kg/h (40.0% and 42.7%) versus propofol (64.0%) required vasopressors during anesthesia [difference and (95% CI) − 24 (− 36.5; − 10.2), − 21.3 (− 34.9; − 7.5)]. Less than 10% of all patients received medications for bradycardia, and < 5% of patients received antihypertensive agents or medications for tachycardia (Table 2).
Subgroup analyses were performed on age, BMI, ASA-PS, amount of remifentanil, duration of IMP administration, cumulative IMP dose, and several combinations. No clinically prominent differences were found in any of these analyses.
Discussion
In this randomized Phase IIb/III trial, the two induction doses of remimazolam (6 and 12 mg/kg/h) showed non-inferiority to propofol (2.0–2.5 mg/kg) in terms of efficacy when used as a sedative for general anesthesia. In fact, the efficacy was 100% in all treatment groups [estimated 95% CI − 0.0487; 0.0250]. Although the incidence of hypotensive events was less in the remimazolam groups compared with the propofol group, the time to extubation was longer in the remimazolam groups compared with the propofol group.
During induction, both doses of remimazolam led to rapid LoC, indicating the ability of the compound to induce anesthesia. Maintenance was assured by continuous IV infusion. The mean (SD) infusion rate during maintenance was 0.97 (0.35) mg/kg/h in the remimazolam 6 mg/kg/h group and 0.99 (0.39) mg/kg/h in the remimazolam 12 mg/kg/h group, resulting in a comparable cumulative dose of remimazolam in both arms. The relatively short and different induction phase in comparison to the maintenance phase with a titration effect is considered responsible for almost identical cumulative doses in both remimazolam groups. Mean BIS values were within the same range in all three groups and indicated a similar and adequate depth of anesthesia. Controllability was rated as “Excellent” or “Good” for 98.7%, 96.7%, and 100% of patients in the remimazolam 6 mg/kg/h, remimazolam 12 mg/kg/h, and propofol groups, respectively.
In terms of recovery, times were significantly shorter among patients in the propofol group (p < 0.05). In addition to the question of whether these absolute differences are clinically meaningful, the use of flumazenil may offer the opportunity to even surpass the recovery speed of propofol.
All treatment regimens were very safe with no deaths during the trial and only two serious but not related AEs in two patients. Overall, a larger percentage of propofol (61.3%) versus remimazolam (41.0%) patients experienced ADRs. Of these, the most frequent ADR was decreased blood pressure in 49.3% of propofol vs. 22% of remimazolam patients. For injection site pain, 18.7% of propofol but no remimazolam patients experienced this ADR. Nausea (7%) and vomiting (6%) in the remimazolam groups were the only ADRs experienced more frequently than in the propofol group (5.3% and 4.0%, respectively). It is also noteworthy that a larger percentage of propofol versus remimazolam patients (64.0% vs 41.3%) required vasopressors and treatment for bradycardia (9.3% vs 6.3%).
Due to the increasing body of evidence of the deleterious effects of intraoperative hypotension, this difference in favor of remimazolam deserves particular attention, although the trial was not fully standardized with regard to concomitant medications and their potential influence on hemodynamics or general fluid management.
The trial could not be fully blinded to investigators, so a theoretical bias can be claimed. However, the primary endpoint consists of composites that are routine in daily practice and thus highly relevant to clinicians. This trial design is deliberately chosen because it comes fairly close to daily clinical practice. It is believed that the majority of anesthesiologists worldwide adjust their medications according to clinical parameters, such as BP and heart rate, and assess the efficacy of anesthetics based on similar parameters. While the authors do acknowledge the lack of standardization in their protocol and the potential influence of repeated rocuronium use during the maintenance phase, they are convinced that the approach taken is a true reflection of the clinical guidance under which the majority of anesthesia is still conducted worldwide. Of note, the BIS values did show an adequate depth of anesthesia in all groups indicating a greater level of objectivity in the results of this trial.
The recovery profile of remimazolam may not have been explored to its full potential due to limited experience with the drug. It is a well-known phenomenon that anesthesiologists need some experience with a new drug before they are able to use the drug to its full advantage and initiate tapering off early enough to allow for the fastest recovery. Therefore, the time intervals during recovery have to be considered with some caution and may not yet represent the full spectrum of remimazolam.
The assessment of intraoperative recall via a post-operative questionnaire (Brice questionnaire) is limited by the fact that remimazolam is expected to have amnestic properties. While it is interesting to determine whether intraoperative awareness has taken place or whether it has happened but simply forgotten, the result and perception are essentially the same for the patient. Therefore, it is believed that the results of the Brice questionnaire, when interpreted as a patient-reported outcome, provide clinically relevant information on intraoperative awareness or recall.
Lastly, the trial design has also not systematically addressed the question of post-operative delirium. This shortcoming is currently being investigated in a European-based Phase 3 trial with remimazolam vs. propofol for general anesthesia (NCT03661489), in which post-operative delirium is being assessed with the NU-DESC in all patients in a systematic manner.
Esterase-metabolized drugs are established pharmacological principles in anesthesia. Remimazolam, a GABA receptorA agonist, transfers this principle to benzodiazepines. This Phase IIb/III trial demonstrated the non-inferiority of remimazolam compared with propofol to induce and maintain general anesthesia in combination with remifentanil and rocuronium. Therefore, remimazolam can be considered a promising alternative for intravenous general anesthesia. Moreover, due to its less pronounced hemodynamic side effects, remimazolam may play an important role in the prevention of intraoperative hypotension. Further clinical trials are needed to demonstrate and better characterize this particular advantage.
References
Chitilian HV, Eckenhoff RG. Anesthetic drug development: novel drugs and new approaches. Surg Neurol Int. 2013;4(Suppl1):S2–S10.
Scott RP, Saunders DA. Propofol: clinical strategies for preventing the pain of injection. Anaesthesia. 1988;43:492–4.
Sneyd JR, Rigby-Jones AE. New drugs and technologies, intravenous anaesthesia is on the move (again). Br J Anaesth. 2010;105:246–54.
Tuk B, van Oostenbruggen MF. Characterization of the pharmacodynamic interaction between parent drug and active metabolite in vivo: midazolam and alpha-OH-midazolam. J Pharmacol Exp Ther. 1999;289:1067–74.
Saari TI, Uusi-Oukari M. Enhancement of GABAergic activity: neuropharmacological effects of benzodiazepines and therapeutic use in anesthesiology. Pharmacol Rev. 2011;63:243–67.
Kilpatrick GJ, McIntyre MS, Cox RF, Stafford JA, Pacofsky GJ, Lovell GG, Wiard RP, Feldman PL, Collins H, Waszczak BL, Tilbrook GS. CNS 7056: a novel ultra-short-acting benzodiazepine. Anesthesiology. 2007;107:60–6.
Schnider T, Minto C. Context sensitive decrement times of remimazolam. Anesth Analg. 2013;117:285.
Upton RN, Somogyi AA. Pharmacokinetics and pharmacodynamics of the short-acting sedative CNS 7056 in sheep. Br J Anaesth. 2010;105:798–809.
Antonik LJ, Goldwater DR. A placebo- and midazolam-controlled phase I single ascending-dose study evaluating the safety, pharmacokinetics, and pharmacodynamics of remimazolam (CNS 7056): part I. Safety, efficacy, and basic pharmacokinetics. Anesth Analg. 2012;115:274–83.
Ihmsen H, Eisenried A. Population pharmacokinetics of remimazolam after continuous infusion in volunteers. Poster 01AP14–1. Euroanaesthesia 2018. Copenhagen, Denmark 2018.
Wiltshire HR, Kilpatrick GJ. A placebo- and midazolam-controlled phase I single ascending-dose study evaluating the safety, pharmacokinetics, and pharmacodynamics of remimazolam (CNS 7056): part II. Population pharmacokinetic and pharmacodynamic modeling and simulation. Anesth Analg. 2012;115:284–96.
Worthington MT, Antonik LJ, Goldwater DR, Lees JP, Wilhelm-Ogunbiyi K, Borkett KM, Mitchell MC. A phase Ib, dose-finding study of multiple doses of remimazolam (CNS 7056) in volunteers undergoing colonoscopy. Anesth Analg. 2013;117:1093–100.
Borkett KM, Riff DS, Schwartz HI, Winkle PJ, Pambianco DJ, Lees JP, Wilhelm-Ogunbiyi K. A Phase IIa, randomized, double-blind study of remimazolam (CNS 7056) versus midazolam for sedation in upper gastrointestinal endoscopy. Anesth Analg. 2015;120:771–80.
Pambianco DJ, Borkett KM, Riff DS, Winkle PJ, Schwartz HI, Melson TI, Wilhelm-Ogunbiyi K. A phase IIb study comparing the safety and efficacy of remimazolam and midazolam in patients undergoing colonoscopy. Gastrointest Endosc. 2016;83:984–92.
Rex DK, Bhandari R, Desta T, DeMicco MP, Schaeffer C, Etzkorn K, Barish CF, Pruitt R, Cash BD, Quirk D, Tiongco F, Sullivan S, Bernstein D. A phase III study evaluating the efficacy and safety of remimazolam (CNS 7056) compared with placebo and midazolam in patients undergoing colonoscopy. Gastrointest Endosc. 2018;88(427–437):e6.
Pastis NJ, Yarmus LB, Schippers F, Ostroff R, Chen A, Akulian J, Wahidi M, Shojaee S, Tanner NT, Callahan SP, Feldman G, Lorch DG Jr, Ndukwu I, Pritchett MA, Silvestri GA, PAION Investigators. Safety and efficacy of remimazolam compared with placebo and midazolam for moderate sedation during bronchoscopy. Chest. 2019;155:137–46.
Doi M. Remimazolam. J Jpn Soc Clin Anesthesia. 2014;34:860–6 (in Japanese, abstract in English).
Probst S, Grossmann E (2014) Phase II study on ultra-short acting remimazolam vs. propofol / sevoflurane in cardiac surgery. Poster presentation. ASA 2014. New Orleans, USA.
International Conference on Harmonisation. Guideline for Good Clinical Practice: E6(R1). International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use ICH Harmonised Tripartite Guideline. Geneva, Switzerland: ICH; 1996:1–59.
World Medical Association. WMA declaration of helsinki—ethical principles for medical research involving human subjects. Fortaleza: WMA; 2013. p. 1–8.
American Society of Anesthesiologists. ASA Physical Status Classification System. 2014.
Johansen JW. Update on bispectral index monitoring. Best Pract Res Clin Anaesthesiol. 2006;20:81–99.
Brice DD, Hetherington RR. A simple study of awareness and dreaming during anaesthesia. Br J Anaesth. 1970;42:535–42.
Acknowledgements
The authors thank Shigehito Sato, who served as the Lead Investigator of this clinical trial of remimazolam at the Hamamatsu University Hospital in Japan. They thank Ichiro Hatono, who provided overall trial monitoring as Director Clinical Research 1st Division, CMIC Co. They thank Kazunori Murakami, who provided data management services at Clinical Information Department, EPS Corporation. They acknowledge Shuya Kiyama, from Jikei University Hospital for serving as Medical Officer and providing advice on the overall conduct of the study and handling of individual subjects for analyses before unblinding. They acknowledge Creative Clinical Research GmbH, Berlin for providing medical writing support in drafting and finalizing this manuscript. They thank Melissa Crawford, PhD, from Edanz Group for editing a draft of this manuscript. This trial was conducted at 49 trial sites in Japan.
Funding
This study is supported by Ono Pharmaceutical Co.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
M. Doi is a consultant of PAION UK and Mundipharma. All others authors have no interests to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article

Cite this article
Doi, M., Morita, K., Takeda, J. et al. Efficacy and safety of remimazolam versus propofol for general anesthesia: a multicenter, single-blind, randomized, parallel-group, phase IIb/III trial. J Anesth 34, 543–553 (2020). https://doi.org/10.1007/s00540-020-02788-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00540-020-02788-6