
Abstract
Purpose
Volatile anesthetic postconditioning reduces apoptosis through antiapoptotic signaling. Whether sevoflurane postconditioning prevents activation of caspase 9 and 3, which are implicated in the initiation and execution step of apoptosis, is not known.
Methods
Isolated perfused guinea pig hearts underwent 30 min global ischemia and 120 min reperfusion [control (CTL)]. Anesthetic postconditioning was elicited with sevoflurane (2%) for 2 min at reperfusion onset (POST). LY294002, phosphatidylinositol-3-kinase (PI3K)/Akt (protein kinase B) inhibitor; and PD98059, extracellular signal-regulated kinase 1/2 (ERK) inhibitor, were administered for 10 min before ischemia and throughout the reperfusion period in POST (POST + LY, POST + PD). Left-ventricular-developed (LVDP) and LV end-diastolic (LVEDP) pressures and infarct size were measured. Western blot analysis determined phosphorylated Akt and ERK expression. Myocardial caspase 3 and 9 were determined immunohistochemically.
Results
After ischemia–reperfusion, POST had higher LVDP (57 ± 9 vs. 38 ± 7 mmHg, p < 0.05) and lower LVEDP (21 ± 8 vs. 46 ± 15 mmHg, p < 0.05) versus CTL. Infarct size was significantly reduced in POST versus CTL (15 ± 3 vs. 41 ± 11%, p < 0.001). Phosphorylation of Akt and ERK after reperfusion was significantly increased in POST versus CTL. Immunoreactivity for caspase 3 and 9 was greater in the nucleus of myocytes and endothelial cells in CTL. POST attenuated this increased immunoreactivity. LY294002 and PD98059 abolished the effect of POST on caspase 3 and 9 immunoreactivity.
Conclusions
Sevoflurane postconditioning prevents activation of caspase 3 and 9, mediators of apoptosis in ischemia–reperfusion injury. This caspase activation is mediated by phosphorylation of Akt and ERK.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myocardial reperfusion is a prerequisite for salvaging ischemic myocardium in acute myocardial infarction. However, restoration of coronary blood flow also paradoxically exacerbates the damage caused by ischemia, termed reperfusion injury. Brief, intermittent episodes of ischemia and reperfusion at the onset of reperfusion after prolonged ischemia confer cardioprotection from reperfusion injury (ischemic postconditioning). Recent studies show that this effect can be induced by brief administration of volatile anesthetics immediately before and after reperfusion, phenomena termed anesthetic preconditioning [1–5] and postconditioning [6, 7]. The mechanisms responsible for anesthetic postconditioning remain unclear. Recent evidence has implicated apoptotic cell death during the reperfusion phase as an important contributor to lethal reperfusion-induced injury [8]. A clinical study has shown that the number of apoptotic cells was greater in postischemic myocardium in patients who received reperfusion treatment than in those who did not [9]. Studies show that the phosphatidylinositol-3-kinase (PI3K)/Akt (protein kinase B) pathway, which is the antiapoptotic prosurvival kinase signaling cascade, plays a pivotal role in reperfusion injury salvage kinase cascade (RISK) pathway in ischemic and anesthetic postconditioning [7, 10–13]. This pathway has been proposed to play a central role in reducing apoptosis by modulating the Bcl-2 family and caspase 9 [14]. Further, a recent study in rat hearts found that sevoflurane postconditioning exerts cardioprotection by activating extracellular signal-regulated kinase 1/2 (ERK) and reducing cytosolic cytochrome C release from mitochondria, which suggests attenuation of apoptosis [15]. It has been reported that ERK activation reduces caspase 3 activity after myocardial ischemia–reperfusion in isolated mouse hearts [16]. Thus, Akt and ERK activation at the time of reperfusion confers cardioprotection against reperfusion-induced injury by reducing apoptosis [17, 18]. Caspase 9 is a critical initiator caspase, and caspase 3 is a terminator caspase, and both are implicated in the execution step of apoptosis [19]. However, no study has demonstrated the effect of sevoflurane postconditioning on immunohistochemical expression of these enzymes in myocardium. The purpose of this study was to examine whether sevoflurane postconditioning prevents caspase 3 and 9 activation and whether Akt and ERK phosphorylation mediate prevention of caspase activation after myocardial ischemia–reperfusion.
Materials and methods
This study was conducted in accordance with the Guidelines for Animal Research at Osaka Dental University and with the approval of the Animal Experiment Committee of Osaka Dental University (No. 09-02044), Osaka, Japan. These guidelines conform to those laid out in the Guide for the Care and Use of Laboratory Animals, available from the National Academy of Science. Male Hartley guinea pigs (Keari, Osaka, Japan) initially weighing 210–260 g were fed Lab Diet guinea pig food (RC4; Oriental Yeast, Tokyo, Japan) and given water ad libitum for 8 weeks.
Isolated heart perfusion and function measurement
Male guinea pigs weighing 650–700 g (12–13 weeks old) were given heparin (1,000 U intraperitoneally), then anesthetized (pentobarbital 60 mg kg−1, intraperitoneally). Hearts were quickly excised and immediately arrested in cold iso-osmotic saline containing 20 mmol L−1 potassium chloride (KCl). The isolated hearts were cannulated via the aorta and perfused at 70 mmHg on a nonrecirculating isovolumic perfused heart apparatus using a Krebs–Henseleit (KH) perfusate (mmol L−1): 118 sodium chloride (NaCl), 4.0 KCl, 2.52 calcium chloride (CaCl2), 24.8 sodium bicarbonate (NaHCO3), 1.7 magnesium sulfate (MgSO4), 1.2 potassium dihydrogen phosphate (KH2PO4), 11.0 glucose, 0.5 ethylenediaminetetraacetic acid (EDTA), and 8 U L−1 insulin. The perfusate was insufflated continuously with 95% oxygen (O2)/5% carbon dioxide (CO2) (pH: 7.41 ± 0.03, partial pressure of oxygen (PO2): 512 ± 10 mmHg, partial pressure of carbon dioxide (PCO2): 39.0 ± 2.2 mmHg) and was filtered through stainless steel membranes with a pore size of 4.0 µm to remove particulate matter. Hearts were paced at 240 beats min−1 using two electroencephalograph (EEG) needle electrodes (NE-224S; Nihon-Kohden, Tokyo) connected to a stimulus generator (SD-5; Grass Instruments, Quincy, MA, USA). Left-ventricular-developed pressure (LVDP; mmHg) was measured using a 2.5-French, Mikro-Tip catheter transducer (SPR-524; Millar Instruments, Inc., Houston, TX, USA) passed into a compliant latex balloon. The LV balloon was connected to a Y-adapter, one end of which was used to advance the micromanometer to the latex balloon. The other end of the Y-adapter was used to fill the LV balloon with bubble-free water to set the LV end-diastolic pressure (LVEDP) at 10 mmHg. LV pressure was recorded on a PowerLab 2/20 data recording system (ADInstruments, Hayward, Australia). The maximum rate LV pressure increase (+dP/dt max) and the minimum rate LV pressure decrease (−dP/dt min) were obtained by electronic differentiation of the LV pressure waveform. Coronary flow (CF) was measured by collecting effluent from the right ventricular outflow tract. Global ischemia was achieved by clamping the aortic inflow line. Ventricular pacing was discontinued during ischemia and was resumed after 2 min of reperfusion. Heart temperature was continuously monitored by a digital thermometer (PTW-100A; Unique Medical, Tokyo, Japan). During ischemia, hearts were maintained at 37°C by enclosure in a water-jacketed air chamber. Warmed perfusate kept in the lower part of the chamber saturated the air with humidity and prevented cooling by evaporation.
Experimental protocol
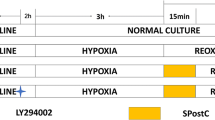
Figure 1 shows the experimental protocol. Animals were randomly assigned to four groups. The hearts were isolated and perfused as described above. After a 20-min equilibration period, baseline LVDP, LVEDP, and CF were recorded, then isolated hearts were subjected to 30 min global ischemia followed by 120 min reperfusion [control group (CTL) n = 8]. Anesthetic postconditioning was elicited sevoflurane administration [2%; one minimum alveolar concentration (MAC)] for 2 min upon reperfusion (POST, n = 8). To investigate the effects of the ERK inhibitor PD98059 (Cayman Chemical, Ann Arbor, MI, USA) and the PI3K inhibitor LY294002 (Enzo Life Sciences International, Plymouth Meeting, PA, USA) on sevoflurane postconditioning and control hearts, PD98059 (20 µmol L−1) and LY294002 (15 µmol L−1) were administered 10 min before ischemia and throughout the ischemia–reperfusion period (POST + PD, POST + LY, CTL + PD and CTL + LY, n = 7 each). Sevoflurane was delivered to the perfusate by bubbling with a 95%O2/5%CO2 gas mixture passing through a calibrated vaporizer (S-MK III; Acoma, Tokyo). Samples of coronary perfusate were collected anaerobically from the aortic cannula for sevoflurane concentration measurement by organic vapor sensor (VOC-101H; O.S.P., Saitama).

Experimental protocol. All hearts were subjected to 30 min global ischemia followed by 120 min reperfusion. Anesthetic postconditioning was elicited by sevoflurane administration [2% or 1 minimum alveolar concentration (MAC)] for 2 min at reperfusion onset (POST). To investigate the effects of PD98059 and LY294002 on sevoflurane postconditioning and control hearts, PD98059 (20 µmol L−1) and LY294002 (15 µmol L−1) were administered 10 min before ischemia and throughout the ischemia–reperfusion periods (POST + PD, POST + LY, CTL + PD and CTL + LY). Tissue samples were obtained at 10 min after reperfusion. CTL control, POST postconditioning, SEVO sevoflurane, PD PD98059 [extracellular signal-regulated kinase 1/2 (ERK) inhibitor], LY LY294002 [phosphatidylinositol-3-kinase (PI3K) inhibitor]
Determination of infarct size
At the end of 120 min reperfusion, the hearts were quickly removed from the Langendorff apparatus and frozen at −80°C for 15 min. They were then sliced into 2-mm-thick transverse sections from apex to base (six slices/heart). After removing the right ventricle and defrosting, each slice was weighed and incubated at 37°C with 1% 2,3,5-triphenyltetrazolium chloride (TTC) (Sigma, St. Louis, MO, USA) in phosphate buffer at pH 7.4 for 10 min and fixed in 10% formalin for at least 5 h to distinguish clearly red-stained viable tissue from pale, unstained, necrotic tissue [20]. Each slice was photographed and the area of necrotic myocardium determined using digital imaging software (Adobe Photoshop CS; Adobe, CA, USA). The area was then multiplied by the weight of the slice and expressed as a fraction of the LV of each heart.
Western blot analysis
To examine Akt and ERK expression and their phosphorylation state over time, separate experiments were performed on four groups of four animals each. For this purpose, the myocardial tissue samples were collected 10 min after reperfusion, frozen in liquid nitrogen, and stored at −80°C until use. Tissue samples were homogenized in ice-cold homogenizing buffer containing in millimolar: 250 sucrose, 20 hydroxyethylpiperazine ethanesulfonic acid (HEPES) (pH 7.5), 10 KCl, 2 ethyleneglycol tetraacetic acid (EGTA), 2 MgCl2, 25 sodium fluoride (NaF), 50 β-glycerophosphate, 1 sodium orthovanadate (Na3VO4), 1 phenylmethylsulphonyl fluoride (PMSF), 1% Triton X, and protease inhibitor leupeptin (10 µg mL−1). The homogenate was centrifuged at 1,000g and 4°C for 5 min to obtain the cytosolic fraction of the tissue. The supernatant containing the cytosolic fraction was centrifuged a second time at 10,000g and 4°C for 15 min.
The protein concentration of the supernatants was estimated using a Bradford assay (Smart Spec 3000; Bio-Rad, Hercules, CA, USA). Bovine serum albumin was used as a standard. Equivalent amounts (50 µg) of protein samples were loaded and separated on a 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (Ready GEL Bio-Rad) and then electrically transferred at 4°C to a polyvinylidene difluoride membrane (Immobilon-P; Millipore, Billerica, MA, USA). Protein transfer was routinely ensured by staining the membrane with Ponceau. After blocking with 5% skim milk in Tris-buffered saline containing 0.1% Tween-20 (TBS-T), the membranes were incubated overnight at 4°C in TBS containing 5% milk and a dilution of primary antibody. The following antibodies were used: phospho-ERK (Thr 202/Tyr 204; 1:500; Cell Signaling Technology, Danvers, MA, USA), ERK (1:1,000; Cell Signaling Technology), phospho-Akt (Ser473; 1:500; Cell Signaling Technology), and Akt (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Membranes were then washed three times with TBS-T for 10 min and subsequently incubated with a 1:1,000 dilution of horseradish peroxidase–labeled antirabbit immunoglobulin G (IgG) (NA 934V; GE Healthcare, Little Chalfont, NA, UK) in TBS-T containing 5% milk. The same blot was stripped and reblotted with antibodies to α-tubulin to confirm equal protein loading. Bound antibody signals were detected with enhanced chemiluminescence (Super Signal; Pierce Biotechnology, Rockford, IL, USA) and visualized using a cooled-CCD imaging system (VersaDoc 5000; Bio-Rad, Hercules, CA, USA). Quantitative analysis of band densities was performed by image analysis software (Quantity One; Bio-Rad).
Immunohistochemistry of activated caspase 3 and 9
Separate experiments were performed to determine immunohistochemical staining for activated caspase 3 and 9 (n = 6 for each group). In this experiment, reperfusion time was extended to 6 h to ensure staining. Infarcted slices were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) for 24 h. At the end of this period, slices were routinely processed and embedded in paraffin. Four-micrometer-thick sections were cut by microtome and deparaffinized in d-limonen for 5 min three times. After rehydrating through a decreasing series of ethanols, sections were washed in distilled water and 50 mM PBS (pH 7.6) for 5 min. They were then treated with 10 mM citrate buffer (pH 6.0) at 121°C for 15 min and washed again with PBS three times. Sections were incubated in a solution of 3% hydrogen peroxide (H2O2) for 5 min to inhibit endogenous peroxidase activity. After this procedure, sections were washed with PBS and incubated with antiactivated caspase 3 (1:100 dilution; Millipore Co, AB3623 South Billerica, MA, USA) and antiactivated caspase 9 (1:100 dilution; Bio Vision, 3149-100, Mountain View, CA, USA) rabbit polyclonal antibodies at 4°C overnight. Next, after washing, sections were incubated with dextran polymer conjugated with horseradish peroxidase (Dako EnVision 2 System HRP) (Dako K4003 DAKO, Carpinteria, CA, USA). Sections were then washed with PBS, incubated with a solution containing 3-3′-diaminobezidine-4HCl (DAB) for 60 s to visualize immunolabeling, and finally counterstained with Mayer’s hematoxylin. Negative controls were treated as above, except incubation with the primary antibody was replaced by incubation with immunoglobulin fraction of normal rabbit. All sections were examined using an Olympus BX50 (Tokyo, Japan) light microscope by two observers blinded to the experimental groups. Five hundred myocytes were counted, and immunohistochemically stained cells were represented as a percentage of total cells [apoptosis index (AI)].
Statistical analysis
Hemodynamic data were tested for normal distribution and subsequently analyzed by a two-factor repeated-measures analysis of variance for time and treatment. If an overall difference between variables was observed, comparisons were performed as one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test for intergroup differences. Analysis of infarct size, immunohistochemical staining, and Western blot was performed using one-way ANOVA followed by Tukey’s post hoc test. A value of p < 0.05 was considered statistically significant (SPSS17 for Windows, SPSS Japan, Tokyo, Japan).
Results
A total of 90 guinea pigs (50 for infarct size study, 16 for Western blot, and 24 for immunohistochemistry) were used. Five hearts were not used secondary to intractable ventricular fibrillation after reperfusion (two in CTL, one in POST, one in CTL + PD, and one in CTL + LY). One heart was not used because of aortic rupture. If a heart was not used, an additional heart was studied. There was no significant difference in body weight among groups. Sevoflurane concentration was 0.30 ± 0.04 mM. Sevoflurane was not detected in effluent during the baseline and ischemic periods nor after discontinuation of sevoflurane during reperfusion. Heart (LV) weights were not different among groups.
Hemodynamics
Baseline LVDP, LVEDP, dP/dt max, dP/dt min and CF were similar among groups (Table 1). Recovery of LVDP was greater in POST compared with CTL throughout the reperfusion period (57 ± 9 vs. 38 ± 7 mmHg, p < 0.05 at 120 min of reperfusion). The improved recovery of LVDP in POST hearts was abolished by administration of PD98059 and LY294002 (POST + PD: 28 ± 14 mmHg, POST + LY: 33 ± 9 mmHg). LVEDP increased significantly compared with baseline in CTL after ischemia–reperfusion. The increase in LVEDP was significantly less in POST compared with CTL hearts during the reperfusion period (21 ± 8 vs. 46 ± 15 mmHg, p < 0.05 at 120 min of reperfusion). LVEDP attenuation increase in POST was lost by PD98059 and LY294002 administration.
All groups had reduced dP/dt max after reperfusion compared with baseline. Recovery of dP/dt min was significantly greater in POST compared with CTL hearts but not in PD98059- and LY294002-treated hearts. Changes of dP/dt min during the reperfusion period were similar to those of dP/dt max. There was no significant difference in CF among groups throughout the experiment. This suggests that CF changes could not account for the improved contractile recovery of POST hearts (Table 1).
Infarct size
Myocardial infarct size in the POST group was significantly reduced by approximately 70% compared with CTL hearts (POST: 15 ± 3% vs. CTL: 41 ± 11%, p < 0.001) (Fig. 2). Myocardial infarct size of the POST group treated with PD98059 and LY294002 did not differ compared with CTL hearts (POST + PD: 43 ± 11%, p = 0.85, POST + LY: 42 ± 6%, p = 0.80 vs. CTL, respectively), suggesting PD98059 and LY294002 abolished the infarct-size-limiting effect of sevoflurane postconditioning. PD98059 and LY294002 did not affect infarct size in CTL (CLT + PD: 42 ± 16%, p = 0.82, CTL + LY: 44 ± 17%, p = 0.65 vs. CTL, respectively).

Infarct size as a percentage of left ventricle (LV). Treatment with sevoflurane (1 MAC) reduced infarct size compared with control (POST: 15 ± 3% vs. CTL: 41 ± 11%, p < 0.001). This cardioprotective effect was abolished by PD98059 and LY294002 (POST + PD: 43 ± 11%, POST + LY: 42 ± 6% vs. CTL, respectively, p = NS). PD98059 and LY294002 did not affect infarct size in CTL. Data are presented as mean ± standard deviation. *p < 0.05: POST versus CTL, POST, POST + PD and POST + LY. CTL control, POST postconditioning, PD PD98059 [extracellular signal-regulated kinase 1/2 (ERK) inhibitor], LY LY294002 [phosphatidylinositol-3-kinase (PI3K) inhibitor]
Western blot analysis
Total ERK and Akt expression and their phosphorylation state, phospho-ERK (Thr 202/Tyr 204) and phospho-Akt (Ser473), 10 min after reperfusion are shown graphically and illustrated by a representative Western blot in Fig. 3. Total and phospho-ERK and Akt densities were normalized against the corresponding CTL densities. Total ERK and Akt expressions were comparable among groups. Phospho-ERK and phosphor-Akt expression were significantly increased in POST compared with CTL. PD98059 and LY294002 administration before ischemia and throughout the reperfusion period abolished increased phosphor-ERK and Akt expression.

Representative Western blot of phosphorylated extracellular signal-regulated kinase 1/2 (phospho-ERK) and phospho-protein kinase B (Akt) from left ventricular samples acquired at 10 min after reperfusion (n = 4 for each group). Phospo-ERK and phospho-Akt expression was significantly increased in POST. PD98059 and LY294002 administration upon reperfusion abolished increased expression of phospo-ERK and phospho-Akt in POST + PD and POST + LY. *p < 0.05: POST versus CTL, POST + PD and POST + LY. CTL control, POST postconditioning, PD PD98059 [extracellular signal-regulated kinase 1/2 (ERK) inhibitor], LY LY294002 [phosphatidylinositol-3-kinase (PI3K) inhibitor]
Immunohistochemistry of active caspase 3 and 9
After 6 h reperfusion, immunohistochemical staining for both caspase 3 and 9 was found in the nucleus of myocytes and endothelial cells in CTL (Figs. 4a, 5a). This immunohistochemical staining for caspase 3 and 9 were attenuated in POST (caspase 3; AI: POST: 2.4 ± 1.3 vs. CTL: 12.3 ± 1.5, p < 0.05, caspase 9; AI: POST: 0.7 ± 0.4 vs. CTL: 12.1 ± 0.9, p < 0.05) (Figs. 4b, 5b and 6). When POST hearts were treated with PD98059 and LY294002, staining for both caspase 3 and 9 was similar to that seen in CTL (caspase 3; AI: POST + PD: 11.5 ± 1.5, POST + LY: 12.3 ± 1.3, caspase 9; AI: POST + PD: 11.8 ± 1.4, POST + LY: 12.5 ± 0.9) (Figs. 4c, d, 5c, d, and 6), suggesting that Akt and ERK are involved in preventing activation of both caspase 3 and 9.

Immunohistochemical staining for activated caspase 3 in the myocardium after 6 h reperfusion. Immunoreactivity was observed in endothelial cells and the nucleus of myocytes (stained brown), which appears morphologically normal in CTL (a). This immunoreactivity was attenuated in POST (b). PD98059 and LY294002 abolished this attenuation (c, d). Original magnification ×200, a CTL, b POST, c POST + PD, d POST + LY. CTL control, POST postconditioning, PD PD98059 [[extracellular signal-regulated kinase 1/2 (ERK) inhibitor], LY LY294002 [phosphatidylinositol-3-kinase (PI3K) inhibitor]

Immunohistochemical staining for activated caspase 9 in the myocardium after 6 h reperfusion. Immunoreactivity was observed in endothelial cells and the nucleus of myocytes (stained brown), which appears morphologically normal in CTL (a). This immunoreactivity was attenuated in POST (b). PD98059 and LY294002 abolished this attenuation (c, d). Original magnification ×200, a CTL, b POST, c POST + PD, d POST + LY. CTL control, POST postconditioning, PD PD98059 [extracellular signal-regulated kinase 1/2 (ERK) inhibitor], LY LY294002 [phosphatidylinositol-3-kinase (PI3K) inhibitor]

Immunohistochemically stained cells as a percentage of 500 cells (apoptosis index). POST reduced the number of stained cells for both activated caspase 3 and 9. This effect was abolished by PD98059 and LY294002. *p < 0.05: POST versus CTL, POST + PD and POST + LY; CTL control, POST postconditioning, PD PD98059 [extracellular signal-regulated kinase 1/2 (ERK) inhibitor], LY LY294002 [phosphatidylinositol-3-kinase (PI3K) inhibitor]
Discussion
This study demonstrated for the first time direct evidence that sevoflurane postconditioning elicited by 1 MAC (2%) for 2 min upon reperfusion reduced the immunohistochemical expression of activated caspase 3 and 9, critical initiator and terminator caspases implicated in the execution step of apoptosis [19]. We also found that phosphorylation of ERK and Akt play a role in preventing caspase 3 and 9 activation. Sevoflurane administration upon reperfusion for only 2 min induced postconditioning. Obal et al. [21] demonstrated that 1 MAC sevoflurane for 2 min immediately after reperfusion is sufficient to protect against myocardial reperfusion injury, and increasing the sevoflurane concentration above 1 MAC resulted in no further protection. Sevoflurane postconditioning has been shown to protect against myocardial reperfusion injury by activating PI3/Akt pathway [12] and ERK [15]. Results of our study are consistent with these studies.
Apoptotic cells are characterized by morphological features, such as chromatin condensation, cytoplasm shrinkage, nuclear fragmentation, and apoptotic bodies in macrophages shown on electron microscopy. Whether apoptosis is truly executed in the experimental model of this study should be determined morphologically. However, these features have been reported to be difficult to observe in the heart [22]. Despite development of several techniques for detecting apoptotic cells their identification remains controversial. The most widely used method is the terminal-transferase-mediated DNA nick-end labeling (TUNEL) staining, which recognizes cells containing DNA fragmentation. However, these DNA strand breaks are not specific for apoptosis but also occur in necrosis [23]. Immunohistochemistry using an antibody to activated (cleaved) caspase 3 has been shown to be a highly specific and sensitive method for detecting apoptosis [9, 24]. In our study, we demonstrated, by using specific antibodies to activated caspase 3 and 9, that sevoflurane postconditioning prevents activation of these enzymes.
It has been demonstrated that cell death due to acute myocardial ischemia occurs predominantly through necrosis and that apoptosis is less important [25]. However, Gottlieb et al. [26] reported that apoptosis after regional ischemia in rabbits hearts is due to reperfusion injury. Mitogen-activated protein kinase/extracellular signal-regulated kinases (MEK) and ERK 1/2 (MEK/ERK) pathway and PI3K/Akt cascade are important signaling pathways that mediate cell survival and cardioprotection [27]. PI3K/Akt pathway activation has been shown to be essential for the antiapoptotic effects of hypoxic preconditioning [28] as well as for infarct size reduction [29]. It has been reported that both Akt and ERK 1/2 are involved in isoflurane-induced anesthetic preconditioning [30, 31]. Recent studies have shown that sevoflurane postconditioning exerts a cardioprotective effect by PI3/Akt signal transduction [12] and by activating ERK and reducing cytosolic cytochrome C release from mitochondria, suggesting attenuation of apoptosis in isolated rat hearts [15].
Caspase 3 is a central effector caspase in many cells, leading to DNase activation followed by DNA fragmentation [32]. Caspase 9 is a critical initiator caspase implicated in the execution step of apoptosis [19]. The importance of apoptosis in cell death following reperfusion has been pharmacologically demonstrated in in vivo rodent models [33, 34]. Activated caspase 3 has been shown to translocate to the nucleus during apoptosis [35] and play an important role in the nuclear changes in apoptotic cells [36]. Many substrates for caspase 3 have been identified in the nucleus [37]. In our study, sevoflurane administration upon reperfusion increased Akt and ERK phosphorylation and attenuated caspase 3 and 9 activation in the nucleus of myocytes and coronary endothelial cells. Attenuation of immunohistochemical staining for activated caspase 3 and 9 in POST was abolished by PD98059 and LY294002, suggesting that preventing both caspase 3 and 9 activation is mediated by ERK and Akt phosphorylation. This is consistent with a previous study suggesting that ERK activation reduces caspase 3 activity after myocardial ischemia–reperfusion in isolated mouse hearts [16].
Akt has been shown to inhibit mitochondria-mediated cell death through phosphorylation of B-cell leukemia 2 family protein antagonist of cell death (BAD) and caspase 9 [38]. Scarabelli et al. [39] demonstrated that endothelial cell apoptosis was mediated completely by caspase 9 activation and that caspase 9 cleavage was observed primarily in endothelial cells using isolated rat hearts subjected to 35 min ischemia followed by 120 min reperfusion. They also showed that different initiator caspases were processed during different phases of ischemia–reperfusion and differentially triggered apoptosis in endothelial cells and cardiomyocytes (caspase 9 for endothelial cells and caspase 8 for cardiomyocytes). Suhara et al. [40] demonstrated that endothelial cells are resistant to apoptosis after death-receptor ligation and caspase 8 activation because they express high levels of FLICE inhibitory protein (FLIP), an endogenous inhibitor of caspase 8 activation. They also showed that PI3K/Akt signaling suppression sensitized endothelial cells to Fas-mediated apoptosis via FLIP regulation. These findings are consistent with our results that attenuation of caspase 9 activation by POST is mediated by PI3K/Akt signaling. In this study, we did not examine the effect of POST on caspase 8. Further research will be needed to elucidate the signaling pathway involved, such as a Fas-mediated pathway.
The following study limitations should be acknowledged. First, we did not confirm direct morphological features of apoptotic cells. Further studies are required to demonstrate apoptotic cells directly. Second, the possibility that sevoflurane postconditioning directly activates antiapoptotic pathways other than MEK/ERK and PI3/Akt cannot be ruled out.
In conclusion, sevoflurane postconditioning prevents activation of caspase 3 and 9, which are mediators of apoptosis in ischemia–reperfusion injury. This caspase activation is mediated by Akt and ERK phosphorylation.
References
Stadnicka A, Marinovic J, Ljubkovic M, Bienengraeber MW, Bosnjak ZJ. Volatile anesthetic-induced cardiac preconditioning. J Anesth. 2007;21:212–9.
Tanaka K, Ludwig LM, Kersten JR, Pagel PS, Warltier DC. Mechanisms of cardioprotection by volatile anesthetics. Anesthesiology. 2004;100:707–21.
Wakeno-Takahashi M, Otani H, Nakao S, Uchiyama Y, Imamura H, Shingu K. Adenosine and a nitric oxide donor enhances cardioprotection by preconditioning with isoflurane through mitochondrial adenosine triphosphate-sensitive K+ channel-dependent and -independent mechanisms. Anesthesiology. 2004;100:515–24.
Kaneda K, Miyamae M, Sugioka S, Okusa C, Inamura Y, Domae N, Kotani J, Figueredo VM. Sevoflurane enhances ethanol-induced cardiac preconditioning through modulation of protein kinase C, mitochondrial KATP channels, and nitric oxide synthase, in guinea pig hearts. Anesth Analg. 2008;106:9–16.
Okusa C, Miyamae M, Sugioka S, Kaneda K, Inamura Y, Onishi A, Domae N, Kotani J, Figueredo VM. Acute memory phase of sevoflurane preconditioning is associated with sustained translocation of protein kinase C-α and ε, but not δ, in isolated guinea pig hearts. Eur J Anaesthesiol. 2009;26:582–8.
Inamura Y, Miyamae M, Sugioka S, Kaneda K, Okusa C, Onishi A, Domae N, Kotani J, Figueredo VM. Aprotinin abolishes sevoflurane postconditioning by inhibiting nitric oxide production and phosphorylation of protein kinase C-δ and glycogen synthase kinase 3β. Anesthesiology. 2009;111(5):1036–43.
Pagel PS. Postconditioning by volatile anesthetics: salvaging ischemic myocardium at reperfusion by activation of prosurvival signaling. J Cardiothorac Vasc Anesth. 2008;22:753–65.
Zhao ZQ, Velez DA, Wang NP, Hewan-Lowe KO, Nakamura M, Guyton RA, Vinten-Johansen J. Progressively developed myocardial apoptotic cell death during late phase of reperfusion. Apoptosis. 2001;6:279–90.
Zidar N, Dolenc-Strazar Z, Jeruc J, Stajer D. Immunohistochemical expression of activated caspase-3 in human myocardial infarction. Virchows Arch. 2006;448:75–9.
Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the reperfusion injury salvage kinase (RISK)-pathway. Cardiovasc Res. 2004;61:448–60.
Tsang A, Hausenloy DJ, Mocanu MM, Yellon DM. Postconditioning: a form of “modified reperfusion” protects the myocardium by activating the phosphatidylinositol 3-kinase-Akt pathway. Circ Res. 2004;95:230–2.
Li H, Wang JK, Zeng YM, Yang CX, Chen HT, Wen XJ, Shui CL, Liang H. Sevoflurane post-conditioning protects against myocardial reperfusion injury by activation of phosphatidylinositol-3-kinase signal transduction. Clin Exp Pharmacol Physiol. 2008;35:1043–51.
Penna C, Perrelli MG, Raimondo S, Tullio F, Merlino A, Moro F, Geuna S, Mancardi D, Pagliaro P. Postconditioning induces an anti-apoptotic effect and preserves mitochondrial integrity in isolated rat hearts. Biochim Biophys Acta. 2009;1787:794–801.
Takatani T, Takahashi K, Uozumi Y, Matsuda T, Ito T, Schaffer SW, Fujio Y, Azuma J. Taurine prevents the ischemia-induced apoptosis in cultured neonatal rat cardiomyocytes through Akt/caspase-9 pathway. Biochem Biophys Res Commun. 2004;316:484–9.
Chen HT, Yang CX, Li H, Zhang CJ, Wen XJ, Zhou J, Fan YL, Huang T, Zeng YM. Cardioprotection of sevoflurane postconditioning by activating extracellular signal-regulated kinase 1/2 in isolated rat hearts. Acta Pharmacol Sin. 2008;29:931–41.
Li DY, Tao L, Liu H, Christopher TA, Lopez BL, Ma XL. Role of ERK1/2 in the anti-apoptotic and cardioprotective effects of nitric oxide after myocardial ischemia and reperfusion. Apoptosis. 2006;11:923–30.
Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia–reperfusion injury in mouse heart. Circulation. 2000;101:660–7.
Matsui T, Tao J, del Monte F, Lee KH, Li L, Picard M, Force TL, Franke TF, Hajjar RJ, Rosenzweig A. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation. 2001;104:330–5.
Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–6.
Fishbein MC, Meerbaum S, Rit J, Lando U, Kanmatsuse K, Mercier JC, Corday E, Ganz W. Early phase acute myocardial infarct size quantification: validation of the triphenyl tetrazolium chloride tissue enzyme staining technique. Am Heart J. 1981;101:593–600.
Obal D, Scharbatke H, Barthel H, Preckel B, Müllenheim J, Schlack W. Cardioprotection against reperfusion injury is maximal with only two minutes of sevoflurane administration in rats. Can J Anaesth. 2003;50:940–5.
Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen M, Voipio-Pulkki LM. Apoptosis in human acute myocardial infarction. Circulation. 1997;95:320–3.
Jugdutt BI, Idikio HA. Apoptosis and oncosis in acute coronary syndromes: assessment and implications. Mol Cell Biochem. 2005;270:177–200.
Gown AM, Willingham MC. Improved detection of apoptotic cells in archival paraffin sections: immunohistochemistry using antibodies to cleaved caspase 3. J Histochem Cytochem. 2002;50:449–54.
Freude B, Masters TN, Robicsek F, Fokin A, Kostin S, Zimmermann R, Ullmann C, Lorenz-Meyer S, Schaper J. Apoptosis is initiated by myocardial ischemia and executed during reperfusion. J Mol Cell Cardiol. 2000;32:197–208.
Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest. 1994;94:1621–8.
Armstrong SC. Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004;61:427–36.
Uchiyama T, Engelman RM, Maulik N, Das DK. Role of Akt signaling in mitochondrial survival pathway triggered by hypoxic preconditioning. Circulation. 2004;109:3042–9.
Murphy E. Primary and secondary signaling pathways in early preconditioning that converge on the mitochondria to produce cardioprotection. Circ Res. 2004;94:7–16.
Raphael J, Rivo J, Gozal Y. Isoflurane-induced myocardial preconditioning is dependent on phosphatidylinositol-3-kinase/Akt signalling. Br J Anaesth. 2005;95:756–63.
Wang C, Weihrauch D, Schwabe DA, Bienengraeber M, Warltier DC, Kersten JR, Pratt PF, Jr., Pagel PS. Extracellular signal-regulated kinases trigger isoflurane preconditioning concomitant with upregulation of hypoxia-inducible factor-1alpha and vascular endothelial growth factor expression in rats. Anesth Analg. 2006;103:281–8. (table of contents).
Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50.
Imamura G, Bertelli AA, Bertelli A, Otani H, Maulik N, Das DK. Pharmacological preconditioning with resveratrol: an insight with iNOS knockout mice. Am J Physiol Heart Circ Physiol. 2002;282:H1996–2003.
Loucks EB, Godin DV, Walley KR, McManus BM, Rahimian R, Granville DJ, Hong JM, Aktary FM, Qayumi AK. Role of platelet activating factor in cardiac dysfunction, apoptosis and nitric oxide synthase mRNA expression in the ischemic-reperfused rabbit heart. Can J Cardiol. 2003;19:267–74.
Kamada S, Kikkawa U, Tsujimoto Y, Hunter T. Nuclear translocation of caspase-3 is dependent on its proteolytic activation and recognition of a substrate-like protein(s). J Biol Chem. 2005;280:857–60.
Zheng TS, Schlosser SF, Dao T, Hingorani R, Crispe IN, Boyer JL, Flavell RA. Caspase-3 controls both cytoplasmic and nuclear events associated with Fas-mediated apoptosis in vivo. Proc Natl Acad Sci USA. 1998;95:13618–23.
Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–6.
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41.
Scarabelli TM, Stephanou A, Pasini E, Comini L, Raddino R, Knight RA, Latchman DS. Different signaling pathways induce apoptosis in endothelial cells and cardiac myocytes during ischemia/reperfusion injury. Circ Res. 2002;90:745–8.
Suhara T, Mano T, Oliveira BE, Walsh K. Phosphatidylinositol 3-kinase/Akt signaling controls endothelial cell sensitivity to Fas-mediated apoptosis via regulation of FLICE-inhibitory protein (FLIP). Circ Res. 2001;89:13–9.
Acknowledgments
We thank Dr. Vincent M. Figueredo for careful review of this manuscript and Dr. Masahiro Wato for his kind advice and help. This study was conducted at the Laboratory Animal Facilities and Dental Bioscience I, Institute of Dental Research, Osaka Dental University, and was supported by Osaka Dental University Research Funds (08-06) (Yoshitaka Inamura) and Grant-in-Aid for Scientific Research (C) 20592382 (Masami Miyamae), “High-Tech Research Center” Project for Private Universities: matching fund subsidy, 2007–2011 from the Ministry of Education, Culture, Sports, Science and Technology of Japan (Naochika Domae).
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Inamura, Y., Miyamae, M., Sugioka, S. et al. Sevoflurane postconditioning prevents activation of caspase 3 and 9 through antiapoptotic signaling after myocardial ischemia–reperfusion. J Anesth 24, 215–224 (2010). https://doi.org/10.1007/s00540-010-0877-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00540-010-0877-6