
Abstract
Background
Previous studies have suggested a link between Helicobacter pylori (H. pylori) infection and nonalcoholic fatty liver disease (NAFLD), yet large-scale longitudinal studies are lacking to elucidate this association.
Methods
A cohort study of 17,028 adults without NAFLD at baseline, who participated in a repeated health screening examination including an H. pylori-specific immunoglobulin G antibody test, was conducted to evaluate the association between H. pylori and NAFLD development. Fatty liver was diagnosed by ultrasonography.
Results
During the 83,130 person-years follow-up, participants with H. pylori infection had a higher rate of incident NAFLD than those who were uninfected. In a multivariable model adjusted for age, sex, body mass index, smoking status, alcohol intake, regular exercise, year of screening exam, and education level, the hazard ratio (HR) for NAFLD development in participants with H. pylori infection compared to those without infection was 1.21 [95% confidence interval (CI), 1.10–1.34]. The association persisted after further adjustment for metabolic variables, inflammatory marker, and liver enzymes. The association between H. pylori and NAFLD was still evident in an analysis using fatty liver index as a surrogate marker of NAFLD. In addition, the association between H. pylori infection and incident NAFLD did not differ across clinically relevant subgroups evaluated.
Conclusions
H. pylori infection was significantly associated with the development of NAFLD, independent of metabolic and inflammatory risk factors. H. pylori infection may play a pathophysiologic role in NAFLD development, indicating that H. pylori eradication might play a role in reducing the risk of NAFLD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Helicobacter pylori (H. pylori) colonizes the stomach of at least half the world’s population and is a key constituent of the human microbiome [1]. Infection is usually acquired early in life and persists throughout the host’s life if untreated [2]. Clinical manifestations of H. pylori infection include peptic ulcer disease, gastric mucosa-associated lymphoid tissue, and non-cardia gastric adenocarcinoma [3]. H. pylori infection not only affects the stomach, but is also linked to a number of extra-gastric diseases, indicating that H. pylori may cause disease by a different biologic process far from the primary site of infection [4]. Notably, a number of studies showed the association between H. pylori infection and cardiovascular disease, type 2 diabetes mellitus, and metabolic syndrome [5–9].
Nonalcoholic fatty liver disease (NAFLD) is characterized by excessive fat infiltration of the liver in absence of significant alcohol intake or secondary causes for steatosis [10]. The clinical consequence of NAFLD is not limited to liver-related morbidity and mortality, but is also associated with cardiovascular disease, type 2 diabetes, and metabolic syndrome [11, 12]. The incidence of NAFLD is rapidly increasing, with huge clinical and economic burdens [13]. Identifying risk factors with potential therapeutic implications is important in managing NAFLD and decreasing these burdens.
Development of NAFLD is a complex process that includes genetic susceptibility and environmental exposures [14]. H. pylori infection has been suggested to play a role in NAFLD development with conflicting results [15–20]. A recent large-scale cross-sectional study of 13,737 adults reported that H. pylori is not associated with NAFLD [21]. In contrast, another study revealed that H. pylori infection was more frequently observed in 28 biopsy-proven NAFLD patients than in 25 healthy controls [19]. Large-scale, longitudinal studies are needed to elucidate the association between H. pylori infection and NAFLD. If proven to be a significant risk factor for NAFLD, H. pylori infection has a therapeutic potential, as it can be eradicated in most patients [22]. Therefore, we longitudinally studied the association between H. pylori infection and NAFLD in a large cohort of asymptomatic men and women.
Methods
Study population
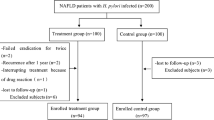
This retrospective cohort study included healthy adults, aged 20 years or older, who participated in a comprehensive health-screening exam at the Center for Health Promotion of the Samsung Medical Center, South Korea, from January 2005 and December 2013 (Fig. 1). Since our objective was to evaluate the longitudinal association between H. pylori infection and NAFLD, we included subjects who underwent at least two screening exams, including abdominal ultrasonography (US), to assess fatty liver status and establish baseline H. pylori infection status (n = 27,914). We excluded 9778 participants who met any of the following exclusion criteria: fatty liver on baseline abdominal US (n = 8305); self-reported history of malignancy (n = 187); self-reported history of chronic liver disease or cirrhosis (n = 138); alcohol intake ≥30 g/day for men and ≥20 g/day for women (n = 2849); positive serologic markers for hepatitis B or C virus (n = 1111). We then excluded 1108 participants with missing data on important covariates, including the following metabolic and inflammatory parameters: 35 missing lipid values, 976 missing homeostasis model assessment of insulin resistance (HOMA-IR) values, 363 missing high-sensitivity C-reactive protein (hsCRP) values. Finally, 17,028 healthy participants without fatty liver at baseline and with available H. pylori status were analyzed. This study was approved by the Institutional Review Board of the Samsung Medical Center, and waived the requirement for informed consent, as we used only de-identified data routinely collected during health screening visits.

Flow diagram of study participants
Data collection
The comprehensive health-screening program included demographic characteristics, anthropometric measurements, detailed physical examination, serum biochemical measurements, and a self-administered health questionnaire on smoking, alcohol consumption, physical activity, medication use, and personal medical history including diabetes mellitus, hypertension, dyslipidemia, and malignancy. Smoking status was categorized as never, former, or current smoker. Alcohol consumption was divided into mild (≤10 g/day) and modest (>10 g/day). Regular exercise was three or more times per week with moderately intense physical activity.
Height and weight were measured to the nearest 0.1 cm and 0.1 kg, respectively, using an Inbody 720 machine (Biospace, Seoul, Korea) while wearing light clothing and with bare feet. Body mass index (BMI) was calculated as weight in kilograms/height in square meters (kg/m2). Blood pressure was measured in the seated position after >5 min of quiet rest using an automated blood pressure monitor (Dinamap PRO 100; GE Healthcare, Milwaukee, WI, USA).
After a ≥12 h fast, blood samples were collected in the morning and analyzed by the hospital clinical laboratory. Serum levels of total cholesterol, triglycerides, low-density lipoprotein cholesterol (LDL-C), and high-density lipoprotein cholesterol (HDL-C) were measured with a Hitachi 7600 (Hitachi, Tokyo, Japan) using enzymatic colorimetric and liquid-selective detergent methods. Serum insulin levels were measured using a radioimmunoassay method with the Packard Cobra II 5010 (Packard Instrument, Baltimore, MD, USA). Serum glucose levels were measured using the hexokinase/glucose-6-phosphate dehydrogenase method with a Hitachi 7600 Modular Dp-110 autoanalyzer (Hitachi, Tokyo, Japan). Serum hsCRP concentrations were measured using an immunoturbidimetric assay (CRPL3, Roche Diagnostics, Indianapolis, IN, USA). The inter- and intra-assay coefficients of variation for quality control specimens were <5% for blood variables. Serum immunoglobulin G (IgG) antibody to H. pylori was measured using an enzyme-linked immunosorbent assay (ELISA) (GAP test IgG kit, Bio-Rad Laboratories Inc., Hercules, CA, USA). Experienced radiologists unaware of the study aims performed the abdominal ultrasonography.
Variables
The primary outcome was incident NAFLD at follow-up, defined by US and exclusion for secondary causes. The US diagnosis of fatty liver was based on standard criteria, including diffuse increased echogenicity of liver parenchyma, liver-to-kidney contrast, deep beam attenuation, and bright vessel walls [23]. Since we excluded participants with excessive alcohol use and/or other chronic liver disease, cases of incident fatty liver were considered NAFLD. Exposure was H. pylori infection status, defined by positive ELISA using serum IgG antibody to H. pylori. Time was calculated from baseline to NAFLD development or last follow-up, whichever came first. For potential confounders, age, sex, BMI, smoking status, alcohol use, regular exercise, year of screening exam, and education level were collected. For potential confounders or mediators, we collected further metabolic and inflammatory variables, and liver enzymes, including aspartate aminotransferase (AST), alanine aminotransferase (ALT), gamma-glutamyl transferase (GGT), hsCRP, fasting blood glucose, triglycerides, LDL-C and HDL-C, and HOMA-IR calculated as [fasting insulin (µU/ml) fasting glucose (mg/dl)]/405 [24].
Statistical analysis
We used Cox regression models to estimate adjusted hazard ratios (aHRs) with 95% confidence intervals (CIs) for incident NAFLD associated with baseline H. pylori seropositivity. Multivariable models were initially adjusted for age (/year) and sex, and then further adjusted for potential confounding factors including year of screening exam, BMI (/kg/m2), smoking status (never vs. past vs. current smoker), alcoholic intake (mild vs. modest), regular exercise (yes vs. no), and education level (elementary school or less vs. middle or high school vs. college or higher). Finally, we fitted additional models adjusted for inflammatory and metabolic factors, and liver function factors that could be potential confounders or mediators of the association between H. pylori infection and incident NAFLD. Model 1 was adjusted for the confounding factors and hsCRP (/mg/dl). Model 2 was further adjusted for HOMA-IR (continuous). Model 3 was adjusted for model 2 plus fasting blood glucose (/mg/dl), triglycerides (/mg/dl), LDL-C (/mg/dl), and HDL-C (/mg/dl). Finally, model 4 was adjusted for model 3 plus AST (/U/l), ALT (/U/l), and GGT (/U/l).
To identify significant factors among the potential mediators, we calculated the mediation effect of metabolic parameters on the association between H. pylori infection and the risk of NAFLD, if metabolic abnormalities met the following three criteria for being a potential mediator: (1) H. pylori infection was associated with metabolic abnormalities, (2) H. pylori infection was significantly associated with the risk of NAFLD, controlling for the metabolic variables, (3) metabolic abnormalities must be significantly related to the risk of NAFLD, and (4) the addition of metabolic variables to the regression model both attenuated the association between H. pylori infection and risk of NAFLD. In addition, we calculated the attenuation of excess risk after adjustment for metabolic variables and inflammatory markers.
We defined the percentage of excess risk mediated (PERM) [25–27] with HRs as follows:
From this equation, we estimated the attenuation or indirect effect of the mediators on NAFLD risk. To estimate the empirical distribution of PERM, we randomly generated 5000 datasets using the bootstrap method and calculated PERM [HR(confounder adjusted), HR(confounder and mediator adjusted)] by fitting Cox proportional hazard models for each dataset. Finally, we used the 50 (median), 2.5, and 97.5th percentiles of the 5000 bootstrap estimates of PERM as the point estimate of PERM and 95% CI.
We conducted subgroup analyses to identify interactions between H. pylori infection and clinically relevant groups, defined by age (<50 vs. ≥50 years), sex (men vs. women), smoking status (non-current vs. current smokers), alcohol intake (mild vs. moderate drinkers), regular exercise (no vs. yes), BMI (<25 vs. ≥25 kg/m2), HOMA-IR (<2.5 vs. ≥2.5), and hsCRP (<0.1 vs. ≥0.1 mg/dl). Interactions between subgroups were tested using likelihood ratio tests comparing models with and without multiplicative interaction terms.
We also examined the association of H. pylori infection with fatty liver index (FLI) as a surrogate marker of NAFLD [28, 29]. The FLI was calculated as follows:
A FLI <30 was considered as indicative of no fatty liver disease, with a FLI ≥30 or ≥60 was considered as indicative of fatty liver disease [30]. We used the same inclusion and exclusion criteria to evaluate the association using FLI, defining fatty liver disease by the FLI rather than abdominal US. Detailed information is provided in Supplementary Materials. A p value <0.05 was considered statistically significant. Statistical analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC, USA).
Results
The mean (SD) age of the 17,028 study participants was 49.3 (9.3) years. Baseline prevalence of H. pylori infection was 58.2% (n = 9918). Participants with H. pylori seropositivity were more likely to be older, male, and current smokers, as well as to have hypertension and higher BMI, total cholesterol, LDL-C, triglycerides, AST, ALT, GGT, and HOMA-IR, a lower education level, and lower levels of HDL-C than those without seropositivity (Table 1). Characteristics of the participants at the end of follow-up are presented in supplementary Table 1.
During the 83,130 person-years follow-up, NAFLD developed in 3381 participants, with an incidence rate of 40.7 per 1000 person-years. The incident rates (per 1000 person-years) of NAFLD by H. pylori serostatus were 37.2 for H. pylori-seropositive participants and 43.2 for those without seropositivity (p < 0.001; Table 2). The median follow-up period was 5.1 years (interquartile range 2.8–7.2). The age- and sex-adjusted HR (95% CI) for incident NAFLD comparing H. pylori-seropositive participants to those without H. pylori seropositivity was 1.14 (1.06–1.22; p < 0.001). The association persisted after further adjustment for year of screening exam, BMI, smoking status, alcohol intake, regular exercise, and education level (HR 1.21, 95% CI 1.10–1.34; p < 0.001). In the multivariable analysis, significant risk factors, other than H. pylori, for NAFLD development were increasing age, BMI, alcohol intake, systolic blood pressure, fasting glucose, LDL-C, triglycerides, AST, ALT, hsCRP, HOMA-IR, and hypoglycemic medication. Protective factors for NAFLD were high education level, HDL-C, and dyslipidemia medication (Table 3).
To explore whether the increased incidence of NAFLD with H. pylori infection was mediated by metabolic risk factors, insulin resistance, inflammatory marker, or liver enzymes, we conducted additional analyses adjusted for potential mediators (Table 4). The association persisted after adjustment for hsCRP as an inflammatory marker (Model 1) or HOMA-IR (Model 2). After adjustment for metabolic variables such as fasting blood glucose, triglycerides, LDL-C, and HDL-C (Model 3), the association between H. pylori infection and incident NAFLD was attenuated but remained statistically significant, suggesting mediation by these metabolic factors.
We calculated the PERM of the inflammatory markers and metabolic variables tested in adjusted models 1, 2, and 3 (Table 5). Among these variables, LDL-C and HDL-C were significant mediators, accounting for 4 and 8% of the total effect of H. pylori infection on NAFLD development, respectively. A combination lipid profile, including LDL-C, HDL-C, and triglycerides, accounted for 11% (4–28) of the excess risk of H. pylori on NAFLD. Dyslipidemia was a partial mediator in the relationship of H. pylori infection and NAFLD, with the HR for NAFLD being reduced in terms of absolute size but remaining different from zero when mediation was introduced. In contrast, hsCRP, fasting blood glucose, and HOMA-IR were not significant mediators for the association.
To evaluate the consistency of the effect of H. pylori on NAFLD, we performed subgroup analyses of factors affecting NAFLD development (Fig. 2). The pre-specified subgroup analysis did not show heterogeneity of risk of incident NAFLD from H. pylori infection, or significant interactions by age (<50 vs. ≥50 years), sex (men vs. women), smoking status (non-current vs. current smokers), alcohol intake (mild vs. moderate drinkers), regular exercise (<3 vs. ≥3 times per week), BMI (<25 vs. ≥25 kg/m2), metabolic syndrome (no vs. yes), HOMA-IR (<2.5 vs. ≥2.5), and hsCRP (<0.1 vs. ≥0.1 mg/dl).

Development of nonalcoholic fatty liver disease (NAFLD) according to H. pylori status in clinically relevant subgroups. aEstimated from Cox proportional hazard models. Multivariable model was adjusted for age, sex, BMI, year of screening exam, smoking status, alcohol intake, regular exercise, and education level. H. pylori Helicobacter pylori, BMI body mass index, HOMA-IR homeostasis model assessment of insulin resistance, hsCRP high-sensitivity C-reactive protein
The association of H. pylori with incident NAFLD was still evident in analysis using the FLI (Supplementary materials and Table 2).
Discussion
In this large cohort study of the association between H. pylori infection and risk of incident NAFLD, we found that participants with H. pylori infection were at higher risk of NAFLD development compared to uninfected individuals. This association persisted even though further adjustment for metabolic risk factors, inflammatory markers, or liver enzymes. Although adjustment for metabolic variables, such as fasting blood glucose, LDL-C, HDL-C, and triglycerides, reduced the association between H. pylori infection and incident NAFLD, the association did remain statistically significant. Among the metabolic variables, lipid metabolism markers, such as LDL-C, HDL-C, and the combination of LDL-C, HDL-C, and triglycerides were significant mediators of the association between H. pylori infection and NAFLD. This association was still evident in the analysis using FLI and was similar across all subgroups. Our findings indicate that H. pylori infection might represent one further hit contributing to NAFLD pathogenesis.
To our knowledge, this is the first large-scale cohort study to evaluate the association between H. pylori infection and the risk of developing NAFLD. H. pylori infection is associated with living in crowded conditions and low socioeconomic status [31, 32]. Metabolic syndrome and NAFLD are usually related to socioeconomic status [33, 34]. We thus conducted this study, paying particular attention to potential confounders, including age, sex, education level, smoking status, alcohol consumption, physical inactivity, and year of screening exam. We also performed further adjustment for metabolic variables and inflammatory markers. Even after adjustment for established NAFLD risk factors and the potential confounders, H. pylori infection was significantly associated with incident NAFLD.
The pathogenetic link between H. pylori infection and NAFLD is debated and clinical data are limited. Several previous studies reported an association between H. pylori infection and NAFLD [6, 16–19, 21]. A recent large-scale cross-sectional study of 13,737 Japanese adults reported that H. pylori infection is not associated with fatty liver disease [21]. Another Korean cross-sectional analysis of 3663 participants found that H. pylori is not a risk factor for NAFLD [18]. In contrast, Polyzos et al. reported that H. pylori infection was more frequently observed in 28 NAFLD patients than in 25 healthy controls [19]. Another study of 130 Japanese participants revealed that the prevalence of nonalcoholic steatohepatitis is higher in H. pylori-infected participants than in non-infected participants [17]. The cross-sectional design of the studies limited assessment of causality; the sample size was small in two studies, which showed positive associations. This large-scale longitudinal study suggests that association between H. pylori and NAFLD is not just an epiphenomenon and H. pylori could be a significant contributor to NAFLD pathogenesis.
Although the exact mechanism linking H. pylori infection and NAFLD needs to be studied in more detail, there are several studies describing a potential role of H. pylori in the predisposition to and causation of NAFLD. H. pylori infection is linked to chronic inflammation, insulin resistance, diabetes, dyslipidemia, and metabolic syndrome [7–9, 16, 35–37]. These variables are also risk factors for NAFLD [38]. In this study, we tested inflammatory and metabolic variables that could be potential mediators linking H. pylori infection and NAFLD. Among these variables, hsCRP, fasting blood glucose, and HOMA-IR were not significant mediators, while lipid markers attenuated the association and accounted for one quarter of the excess risk of H. pylori on NAFLD. These findings suggest that H. pylori infection contributes to development NAFLD by altered lipid metabolism. NAFLD is closely linked to abnormal lipid metabolism, resulting in increased hepatic accumulation of triacylglycerol [39]. Notably, H. pylori infection can alter lipid profiles, including low HDL-C, high LDL-C and triglycerides [8, 9, 40]. The unexplained risk by the metabolic mediators might be caused by other mechanisms such as H. pylori induced gastric atrophy leading to loss of acid, subsequent small intestinal bacterial overgrowth (SIBO), increased intestinal permeability, and increased portal endotoxins [15, 41]. It is reported that SIBO may reflect qualitative and quantitative changes in the microbiota, leading to intestinal barrier disruption, subsequent bacterial translocation, and development of portal endotoxemia. As a result, endotoxin-mediated cytokines were increased in the liver and enhanced hepatic inflammation and fibrosis [42, 43]. Future studies will be required to evaluate this possibility.
Several limitations need to be considered in interpreting our findings. First, we defined NAFLD by US after exclusion of secondary causes for steatosis. US may lead to an incorrect diagnosis of NAFLD in 10 to 30% of cases [44]. However, many population-based studies have relied on US to diagnose fatty liver [45, 46] and the results did not differ when fatty liver was defined using the FLI. Second, H. pylori infection status was evaluated solely with serum IgG to H. pylori measured by ELISA, without other laboratory assessments, such as a rapid urease test or urease breath test. Accordingly, the serologic test cannot discriminate accurately between current and past infections. This may not necessarily be a disadvantage, as past infection may even be more relevant for disease pathogenesis. In addition, the serum IgG antibody test to H. pylori is inexpensive mass-screening tool that can be used easily in areas with a high prevalence of H. pylori infection. Prevalence of H. pylori infection in Korea from a nationwide multicenter study was 59.6%; this result did not differ from the 58.2% in our data [47, 48]. Third, we did not have the information of H. pylori eradication. Effect of H. pylori eradication on the risk of NAFLD cannot be assessed and the results might have been influenced by H. pylori eradication. However, in Korea, according to our national health policy and guidelines, indications for H. pylori eradication include peptic ulcer disease, infection after endoscopic resection for early gastric cancer and gastric mucosa-associated lymphoid tissue [49, 50]. Accordingly, we expected that most asymptomatic participants with H. pylori seropositivity had not received medication of H. pylori eradication. Also, we excluded participants who were diagnosed with gastric malignancy in this study. Finally, this study focused on healthy men and women who underwent routine health check-ups; thus, it may be difficult to generalize our findings to other populations.
This study is strengthened by the cohort design used, the relatively large sample size and exclusion of baseline NAFLD, which allowed us to identify causal associations. This is usually impossible in cross-sectional studies. Additional strengths include the use of high-quality, standardized clinical and laboratory methods.
In conclusion, this study showed that H. pylori infection was associated with an increased risk of NAFLD development in asymptomatic participants without baseline NAFLD. This effect might be exerted through markers of lipid metabolism. These findings support the hypothesis that H. pylori may contribute to the pathogenesis of NAFLD. Further investigations are needed to determine whether H. pylori eradication can improve NAFLD risk.
References
Everhart JE. Recent developments in the epidemiology of Helicobacter pylori. Gastroenterol Clin North Am. 2000;29:559–78.
Ernst PB, Peura DA, Crowe SE. The translation of Helicobacter pylori basic research to patient care. Gastroenterology. 2006;130:188–206.
Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med. 2002;347:1175–86.
Goni E, Franceschi F. Helicobacter pylori and extragastric diseases. Helicobacter. 2016;21(Suppl 1):45–8.
Park MJ, Choi SH, Kim D, et al. Association between Helicobacter pylori seropositivity and the coronary artery calcium score in a screening population. Gut Liver. 2011;5:321–7.
Jamali R, Mofid A, Vahedi H, et al. The effect of Helicobacter pylori eradication on liver fat content in subjects with non-alcoholic fatty liver disease: a randomized open-label clinical trial. Hepat Mon. 2013;13:e14679.
Hsieh MC, Wang SS, Hsieh YT, et al. Helicobacter pylori infection associated with high HbA1c and type 2 diabetes. Eur J Clin Invest. 2013;43:949–56.
Satoh H, Saijo Y, Yoshioka E, et al. Helicobacter pylori infection is a significant risk for modified lipid profile in Japanese male subjects. J Atheroscler Thromb. 2010;17:1041–8.
Gunji T, Matsuhashi N, Sato H, et al. Helicobacter pylori infection is significantly associated with metabolic syndrome in the Japanese population. Am J Gastroenterol. 2008;103:3005–10.
Korean Association for the Study of the L. KASL clinical practice guidelines: management of nonalcoholic fatty liver disease. Clin Mol Hepatol. 2013;19:325–48.
Adams LA, Waters OR, Knuiman MW, et al. NAFLD as a risk factor for the development of diabetes and the metabolic syndrome: an eleven-year follow-up study. Am J Gastroenterol. 2009;104:861–7.
Lonardo A, Loria P. NAFLD and cardiovascular risk: direct evidence for the tale of two ages. Am J Gastroenterol. 2009;104:1851–2.
Younossi ZM, Koenig AB, Abdelatif D, et al. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84.
Sookoian S, Pirola CJ. Nonalcoholic fatty liver disease and metabolic syndrome: shared genetic basis of pathogenesis. Hepatology. 2016;64:1417–20.
Li M, Shen Z, Li YM. Potential role of Helicobacter pylori infection in nonalcoholic fatty liver disease. World J Gastroenterol. 2013;19:7024–31.
Polyzos SA, Kountouras J, Zavos C, et al. Helicobacter pylori infection, insulin resistance and nonalcoholic fatty liver disease. Med Hypotheses. 2014;82:795.
Sumida Y, Kanemasa K, Imai S, et al. Helicobacter pylori infection might have a potential role in hepatocyte ballooning in nonalcoholic fatty liver disease. J Gastroenterol. 2015;50:996–1004.
Baeg MK, Yoon SK, Ko SH, et al. Helicobacter pylori infection is not associated with nonalcoholic fatty liver disease. World J Gastroenterol. 2016;22:2592–600.
Polyzos SA, Kountouras J, Papatheodorou A, et al. Helicobacter pylori infection in patients with nonalcoholic fatty liver disease. Metabolism. 2013;62:121–6.
Takuma Y. Helicobacter pylori infection and liver diseases. Gan To Kagaku Ryoho. 2011;38:362–4.
Okushin K, Takahashi Y, Yamamichi N, et al. Helicobacter pylori infection is not associated with fatty liver disease including non-alcoholic fatty liver disease: a large-scale cross-sectional study in Japan. BMC Gastroenterol. 2015;15:25.
Gatta L, Vakil N, Vaira D, et al. Global eradication rates for Helicobacter pylori infection: systematic review and meta-analysis of sequential therapy. BMJ. 2013;347:f4587.
Saverymuttu SH, Joseph AE, Maxwell JD. Ultrasound scanning in the detection of hepatic fibrosis and steatosis. Br Med J (Clin Res Ed). 1986;292:13–5.
Matthews DR, Hosker JP, Rudenski AS, et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–9.
Global Burden of Metabolic Risk Factors for Chronic Diseases C, Lu Y, Hajifathalian K, Ezzati M, et al. Metabolic mediators of the effects of body-mass index, overweight, and obesity on coronary heart disease and stroke: a pooled analysis of 97 prospective cohorts with 1.8 million participants. Lancet. 2014;383:970–83.
Lin DY, Fleming TR, De Gruttola V. Estimating the proportion of treatment effect explained by a surrogate marker. Stat Med. 1997;16:1515–27.
Hafeman DM. ”Proportion explained”: a causal interpretation for standard measures of indirect effect? Am J Epidemiol. 2009;170:1443–8.
Kim JH, Kwon SY, Lee SW, et al. Validation of fatty liver index and lipid accumulation product for predicting fatty liver in Korean population. Liver Int. 2011;31:1600–1.
Meffert PJ, Baumeister SE, Lerch MM, et al. Development, external validation, and comparative assessment of a new diagnostic score for hepatic steatosis. Am J Gastroenterol. 2014;109:1404–14.
Bedogni G, Bellentani S, Miglioli L, et al. The fatty liver index: a simple and accurate predictor of hepatic steatosis in the general population. BMC Gastroenterol. 2006;6:33.
Malaty HM, Graham DY. Importance of childhood socioeconomic status on the current prevalence of Helicobacter pylori infection. Gut. 1994;35:742–5.
Moayyedi P, Axon AT, Feltbower R, et al. Relation of adult lifestyle and socioeconomic factors to the prevalence of Helicobacter pylori infection. Int J Epidemiol. 2002;31:624–31.
Park MJ, Yun KE, Lee GE, et al. A cross-sectional study of socioeconomic status and the metabolic syndrome in Korean adults. Ann Epidemiol. 2007;17:320–6.
Jia G, Li X, Wang L, et al. Relationship of socioeconomic status and non-alcoholic fatty liver disease in patients with type 2 diabetes mellitus. Zhonghua Gan Zang Bing Za Zhi. 2015;23:760–4.
Polyzos SA, Kountouras J, Zavos C, et al. The association between Helicobacter pylori infection and insulin resistance: a systematic review. Helicobacter. 2011;16:79–88.
Peach HG, Barnett NE. Helicobacter pylori infection and fasting plasma glucose concentration. J Clin Pathol. 2001;54:466–9.
Gillum RF. Infection with Helicobacter pylori, coronary heart disease, cardiovascular risk factors, and systemic inflammation: the third national health and nutrition examination survey. J Natl Med Assoc. 2004;96:1470–6.
Chitturi S, Abeygunasekera S, Farrell GC, et al. NASH and insulin resistance: insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology. 2002;35:373–9.
Puri P, Baillie RA, Wiest MM, et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007;46:1081–90.
Kim TJ, Lee H, Kang M, et al. Helicobacter pylori is associated with dyslipidemia but not with other risk factors of cardiovascular disease. Sci Rep. 2016;6:38015.
Miele L, Valenza V, La Torre G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–87.
Korponay-Szabo IR, Halttunen T, Szalai Z, et al. In vivo targeting of intestinal and extraintestinal transglutaminase 2 by coeliac autoantibodies. Gut. 2004;53:641–8.
Sandler NG, Koh C, Roque A, et al. Host response to translocated microbial products predicts outcomes of patients with HBV or HCV infection. Gastroenterology. 2011;141:1220–30.
Hernaez R, Lazo M, Bonekamp S, et al. Diagnostic accuracy and reliability of ultrasonography for the detection of fatty liver: a meta-analysis. Hepatology. 2011;54:1082–90.
Clark JM, Diehl AM. Defining nonalcoholic fatty liver disease: implications for epidemiologic studies. Gastroenterology. 2003;124:248–50.
Lazo M, Hernaez R, Eberhardt MS, et al. Prevalence of nonalcoholic fatty liver disease in the United States: the third national health and nutrition examination survey, 1988–1994. Am J Epidemiol. 2013;178:38–45.
Lim SH, Kwon JW, Kim N, et al. Prevalence and risk factors of Helicobacter pylori infection in Korea: nationwide multicenter study over 13 years. BMC Gastroenterol. 2013;13:104.
Yim JY, Kim N, Choi SH, et al. Seroprevalence of Helicobacter pylori in South Korea. Helicobacter. 2007;12:333–40.
Kim SG, Jung HK, Lee HL, et al. Guidelines for the diagnosis and treatment of Helicobacter pylori infection in Korea, 2013 revised edition. Korean J Gastroenterol. 2013;62:3–26.
Kim N, Kim JJ, Choe YH, et al. Diagnosis and treatment guidelines for Helicobacter pylori infection in Korea. Korean J Gastroenterol. 2009;54:269–78.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kim, T.J., Sinn, D.H., Min, Y.W. et al. A cohort study on Helicobacter pylori infection associated with nonalcoholic fatty liver disease. J Gastroenterol 52, 1201–1210 (2017). https://doi.org/10.1007/s00535-017-1337-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-017-1337-y