
Abstract
Background
Epidemiological studies demonstrate a link between gastrointestinal cancers and environmental factors such as diet. It has been suggested that environmental cancer risk is determined by the interaction between diet and microbes. Thus, the purpose of this study was to examine the hypothesis that microbiota composition during colorectal cancer (CRC) progression might differ depending on the stage of the disease.
Methods
A total of 28 age-matched and sex-matched subjects, seven with CRC adenocarcinoma, 11 with tubular adenomas and ten healthy subjects with intact colon, were included into the study. Microbiomes from mucosal and fecal samples were analyzed with 16S ribosomal RNA gene pyrosequencing, together with quantitative PCR of specific bacteria and archaea.
Results
The principal coordinates analysis clearly separated healthy tissue samples from polyps and tumors, supporting the presence of specific bacterial consortia that are associated with affected sites and that can serve as potential biomarkers of CRC progression. A higher presence of Fusobacterium nucleatum and Enterobacteriaceae was found by qPCR in samples from CRC compared to healthy controls. We observed a correlation between CRC process development and levels of Methanobacteriales (R = 0.537, P = 0.007) and Methanobrevibacterium (R = 0.574, P = 0.03) in fecal samples.
Conclusion
Differences in microbial and archaeal composition between mucosal samples from healthy and disease tissues were observed in tubular adenoma and adenocarcinoma. In addition, microbiota from mucosal samples represented the underlying dysbiosis, whereas fecal samples seem not to be appropriate to detect shifts in microbial composition. CRC risk is influenced by microbial composition, showing differences according to disease progression step and tumor severity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is the most common malignancy in the Western World, and it also constitutes the second most important cause of cancer death, with an overall 5-year survival rate of 64 % [1, 2].
Colorectal cancer is a multifactorial disease and the etiology is complex. The majority of CRC occurrences are considered sporadic, although there is an important genetic influence in the development of CRCs. There are certain special risk groups, including Lynch syndrome, familial adenomatous polyposis, family and personal history of CRC, some cases of chronic inflammatory bowel disease, presence of adenomatous colon polyps and age over 50 years. However, environmental factors including diet and lifestyle and also microbiota are considered to impact on CRC occurrence [3, 4]. Within the human colon, up to one hundred trillion bacteria coexist in commensal balance in healthy individuals, playing a crucial role in food digestion, protection of intestinal mucosa, and modulation of the immune system [5]. It has been suggested that the interaction between diet and microbiota metabolism may impact on the environmental cancer risk [6]. Because of the preponderant role of intestinal microbiota in gut development and pathology, especially inflammatory bowel disease (IBD) as a risk factor for CRC [7], early efforts have focused on elucidating the multifactorial role of gut microbes in CRC [8]. More recently, many efforts have been made to unveil the contribution of intestinal microbiota to gut diseases, employing culture-independent techniques. Alterations in microbial community structure influence the health condition of the colon, as has been shown, both in humans and animal models [2, 3, 9, 10].
There is evidence of microbial dysbiosis in colorectal cancer patients, particularly when comparing between CRC tissue and adjacent non-malignant mucosa [11], as well as in inflammation-induced tumor progression of some polyps to cancer [12]. An increase in Bacteroides-Prevotella group in CRC mucosa was described [13], whereas another report identified shifts involving more than one genus in cancer patients [14]. Differences in fecal microbiota have also been reported between CRC and patients with colonic polyps compared to controls, with a significant increase of two Clostridium species [15].
In contrast to gastric cancer, where just one bacteria (Helicobacter pylori) has been associated to the disease, many pathogenic species have been assessed as directly responsible in 15 % of the total CRC cases [16], and among them Fusobacterium nucleatum has been found to be strongly prevalent [17]. Akkermansia muciniphila, a new mucin-degrading bacterium, might also be involved in the formation of an inflammatory-driven colonic disease [18]. Additionally, little is known about the relationship between archaea methanogens species and different colonic diseases, particularly with CRC [19].
It has been suggested that CRC disease develops over time as sequential multi-step process, known as the “adenoma-carcinoma sequence” [20]. Thus, we hypothesized that microbiota composition during cancer progression might be different depending on the status of the disease, and we determine for the first time microbial composition (both bacteria and archaea) in tumors, colonic polyps and healthy sites, in many cases from the same individuals. We compare the microbial population in mucosa samples of CRC patients with tubular adenomas and adenocarcinoma vs. healthy controls. Tubular adenomas are usually removed because of their high tendency to become malignant and to lead to colon cancer development. We also evaluated bacterial diversity and composition in fecal samples of the same patients, in order to study the potential development of non-invasive diagnostic tools. We studied microbial composition with two different approaches: a species-specific quantification of several bacterial and archaeal micro-organisms (by real-time qPCR) that have been associated to the disease; and an open-ended, high-throughput estimate of bacterial diversity (by 16S rRNA pyrosequencing), in which all genera potentially associated with colonic polyps, tumors or health conditions can be identified.
Methods
Subjects and samples collection
Subjects were selected randomly from the Gastroenterology Unit of the Hospital Universitario Central of Asturias (HUCA) in Oviedo, Asturias (Spain). A written informed consent was obtained from the participants and the study protocol was approved by the Principality of Asturias Clinical Research Ethical Committee, Spain. All cases were adults who underwent ambulatory colonoscopy for any indication, and in whom neoplastic polyps or colorectal cancer were detected. We used asymptomatic patients (61.43 ± 14.7 years old) included in a program of screening for familial first-degree colorectal cancer as controls; and excluded patients with hereditary syndromes (familial adenomatous polyposis [FAP] and hereditary non-polypoid colorectal cancer [HNPCC]) and with positive familial antecedents of CRC.
For this study, stool samples and colorectal biopsies from patients were analyzed. Fecal samples were obtained at least 1 week before to colonoscopy; these were immediately frozen and stored at −80 °C until microbiological analysis. Total colonoscopy was performed under i.v. sedation in both patients and controls, and they signed a detailed informed consent before the procedure. The method used for bowel cleaning for all the participants was as follows: (a) low-fiber diet for 72 h before the colonoscopy; (b) full liquid diet during the day before the colonoscopy; (c) ingestion of two tablets of bisacodyl–5 mg, the day before the colonoscopy around 7 p.m.; (d) drinking of 1 l of polyethylene glycol + ascorbic acid (Moviprep™) at 8 p.m. on the day before and repeated at the same dose and quantity at 6 a.m. the same day of the colonoscopy. The procedure was done in the morning or afternoon, between 9 and 14 h.
Biopsy samples were taken from the normal mucosa (rectum) in controls, and from the patients (polyps and tumor). Two biopsies for histopathological analysis (Table 1) were obtained, and two more for microbiological studies (one from lesion parts and other one from normal mucosa adjacent to the lesions); these were immediately frozen and stored at −80 ºC, until their analysis.
DNA extraction
DNA extraction from fecal samples (220–250 mg) and mucosal tissue (5–15 mg) was performed using commercial kits (Macherey–Nagel, Germany) according to the manufacturer’s instructions, with a previous glass bead beating (0.17 mm diameter). DNA concentration and integrity were measured by calculating A260/280 ratios in a Quawell Spectrophotometer (Thermo Fisher Scientific).
16S rRNA gene pyrosequencing
The first 500 bp of the 16S rRNA genes were amplified with the universal eubacterial primers 27F and 533R, using the high-fidelity AB-Gene DNA polymerase (Thermo Scientific) with an annealing temperature of 52 °C and 20 cycles to minimize PCR biases [21]. The universal primers were modified to contain the pyrosequencing adaptors A and B and an 8-bp “barcode” specific to each sample. Barcodes were different from each other in at least three nucleotides to avoid mistakes in sample assignments. Two PCRs were used per sample, pooling their PCR products before purification, which was performed using an Nucleofast PCR purification kit (Macherey–Nagel), and further cleaned by AMPure XP beads (Roche, Basel, Switzerland) before pyrosequencing. The final DNA concentration per sample was measured by picogreen fluorescence in a Modulus 9200 fluorimeter from Turner Biosystems, and samples were mixed in equimolar amounts in groups of 20. PCR products were pyrosequenced from the forward primer end only in one-eighth of pyrosequencing plates using a GS-FLX sequencer with Titanium chemistry (Roche) at the Center for Public Health Research (CSISP-FISABIO) in Valencia, Spain.
Sequence analysis
Reads with an average quality value lower than 20 and/or with more than our ambiguities in homopolymeric regions in the first 360 flows were excluded from the analysis. Only reads longer than 200 bp were considered, and chimeric sequences were filtered out using Mothur [22]. The ends of reads were found to have an increased rate of ambiguous base calls, and sequences were therefore end-trimmed in 10-bp windows of mean quality values <20 using Prinseq. Sequences were assigned to each sample by the 8-bp barcode and passed through the ribosomal database project classifier [23], where each read was assigned a phylum, class, family and genus, as long as the taxonomic assignment was unambiguous within an 80 % confidence threshold. To estimate total diversity, sequences were clustered at 97 % nucleotide identity over 90 % sequence alignment length using the RDP pyrosequencing pipeline, and rarefaction curves obtained with the program analytic rarefaction 1.3. For this analysis, sequences over 97 % identical were considered to correspond to the same operational taxonomic unit (OTUs), representing a group of reads that likely belong to the same species [24]. Principal coordinates analyses (PCoA) were performed with UNIFRAC [25] using clustering at 97 % sequence identity. The Unifrac analysis compares the 16S-estimated diversity with a phylogenetic approach that takes into account both taxonomically assigned and unassigned reads.
Bacterial analysis using real-time quantitative PCR
PCR primers were targeted to total bacteria count and also to Bifidobacterium spp., Lactobacillus spp.; Bacteroides-Prevotella group; Enterobacteriaceae family; Blautia coccoides group; Akkermansia muciniphila; Faecalibacterium prausnitzii and Fusobacterium nucleatum (Table S1). The qPCRs were conducted as previously described [26]. The qPCR amplification and detection were performed in a LightCycler® 480 Real-Time PCR System (Roche). Each reaction mixture of 10 μl was composed of SYBR® Green PCR Master Mix (Roche), 0.5 μl of each of the specific primers at a concentration of 0.25 μM, and 1 μl of template DNA. The PCR products were detected in the last step of each cycle. A melting curve test was made after amplification to distinguish the targeted from the non-targeted PCR product. The bacterial concentration in each sample was calculated comparing the Ct values obtained from standard curves. These were created using serial tenfold dilution of pure culture-specific DNA fragments corresponding to 10–109 gene copies/ml.
Archaea analysis using real-time quantitative PCR
Quantitative real-time PCR was performed using ABI Prism 7900HT (Applied Biosystems) with four different primers and probe sets (Table S1): two hidrogenotrophic orders (methanomicrobiales (MMB) and methanobacteriales (MBT)) and two acetoclastic families (methanosaetaceae (Mst) and methanosarcinaceae (Msc)). Each reaction mixture was carried out in a total volume of 10 µl containing genomic DNA, 1X Taqman universal PCR Master Mix (Applied Biosystems), 0.9 μM of forward and reverse primer and 0.2 μM of the probe. The Taqman Probes were labeled with the fluorescent dyes FAM (reporter) and TAMRA (quencher). We also analysed the presence of Methanobrevibacter genus (MET) and Methanobrevibacter smithii (Mnif) by specific-qPCR in a LightCycler® 480 Real-Time PCR System (Roche) using SYBR® Green PCR Master Mix (Roche). Each reaction mixture of 10 μl was composed of 0.5 μl of each of the specific primers at a concentration of 0.25 μM, and 1 μl of template DNA.
All amplifications were performed in triplicate and a corresponding standard curve was included. Triplicate results of the qPCR were averaged and the standard deviation calculated. The thermal cycling protocol was as follows: initial denaturation of 10 min at 95 °C, followed by 45 °C denaturation cycles at 95 °C for 15 s, annealing at 50 °C (Mst-set, Msc-set) or 54 °C (MBT-set) or 55 °C (MET and Mnif) for 30 s and extension at 72 °C for 30 s. For the MMB primer set, the amplification was carried out using a two-step thermal cycling protocol consisting of 45 cycles of 15 s at 95 °C and 90 s at 63 °C.
Standard curves were generated using a plasmid containing the full-length 16S rRNA gene sequences from the representative strains of the target methanogenic group, as described previously [27]. Genomic DNAs from four archaea (Methanobacterium formicicum DSM 1535; Methanoculleus bourgensis DSM 3045; Methenosarcina barkeri DSM 800 and Methanosaeta concilii DSM 2139; Methanobrevibacterium smithii DSM 2374) were used for standard curves. DNA concentration was measured with the Picogreen double-stranded DNA quantification kit (Invitrogen) and a tenfold serial dilution series (102–109 copies) was generated for each standard curve and analyzed by quantitative PCR in triplicate with its corresponding primer and probe set.
qPCR statistical analysis
For statistical analysis, SPSS 17.0 software was used. Due to the non-normal distribution of microbial data, they were expressed as medians with interquartile ranges (IQR) and non-parametric tests were performed. Friedman’s test was used to compare microbial groups through time (paired samples). Mann–Whitney U test was used for comparisons between two groups. For comparisons of more than two groups, Kruskal–Wallis test was applied, and statistical differences were corrected for a multiple comparison test using the Bonferroni correction. The χ 2 test was applied to establish differences in bacterial prevalence between the studied groups. A P value <0.05 was considered statistically significant. Spearman rank test allowed the study of the correlation between variables, and significance was established at a coefficient of 0.5 %.
Results
Eligible subjects and samples
A total of 28 subjects were included in the study. From those, seven corresponded to the CRC group, 11 to the tubular adenoma (polyp) group and ten healthy individuals to the control group. There were seven males in the CRC group, and ten males and one female in the polyp group. In the healthy group, six males and four females were included. The mean (±SD) age of the subjects was 71.1 ± 10.1 years for the CRC group and 63.3 ± 13.1 years for the polyp group, whereas the age of the healthy group was 52.6 ± 15.2 years. One biopsy and one fecal sample were collected from each patient and each healthy subject, with some exceptions. Finally, we collected four complete sets of samples for the healthy group, six for the polyp group and seven for the adenocarcinoma group. Patient characteristics and details of the surgical treatment performed are shown in Table 1.
Mucosal and fecal microbiota composition by 16S rRNA gene pyrosequencing
After sequence length and quality filtering, we obtained an average of 3,494 reads of the 16S rRNA gene per sample. Sequences are available in the MG-RAST server with accession numbers 4542409.3 through 4542468.3, under the project name “Colorectal Cancer Microbiota”. The 16S rRNA reads were grouped by sample type, and the number of sequence clusters at 97 % of sequence identity was used as an estimate of the approximate number of species, given that a 3 % of divergence in this gene has been established as the threshold for prokaryotic species boundaries [24]. The number of species-level “operational taxonomic units” (OTUs) was related to the sequencing effort in rarefaction curves, where the level of bacterial diversity can be compared between sample types (Fig. 1). Tissue samples were found to have almost half of the diversity observed in fecal samples (Fig. 1a). This could be the outcome of fecal material gathering microbial cells from the whole gastrointestinal tract, whereas biopsies are more specific samples that exclusively represent the microbiota at the sampling site. Thus, feces are likely to contain a large number of microbial species unrelated to the disease site, which may introduce noise in the detection of potential biomarkers of the disease.

Microbial diversity of mucosal tissue (biopsies) and intestinal content (feces). Graphs show rarefaction curves by estimating of the number of bacterial species, as inferred by the number of operational taxonomic units (OTUs). An OTU was a cluster of 16S rRNA sequences that were over 95 % identical, a conservative estimate for the boundary between species, established at 97 % for full-length 16S gene sequences. a shows rarefaction curves for biopsies and fecal samples. b shows rarefaction curves for the biopsies from the three groups (control, polyp and tumor)
When the different biopsies were analyzed, tumor samples displayed the highest level of diversity, having an estimated number of species 75 % higher than tissues from healthy sites, which were used as a control (Fig. 1b). Polyp samples displayed intermediate diversity values.
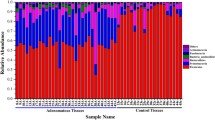
The bacterial compositions of different samples, as estimated by assignment of the 16S rRNA reads to the RDP database, show that feces and biopsies contain very different bacterial communities, with some genera like Bacillus or Staphylococcus being absent in fecal material (Fig. 2). In addition, all three fecal samples (from individuals having polyps or tumors, as well as from healthy controls) showed a similar bacterial composition at the genus taxonomic level. Tissue samples did show differences between healthy and affected tissues with several genera, like Blautia and Prevotella, which were virtually absent in healthy tissues but present in similar proportion in polyps and tumors, and could therefore be used as potential biomarkers of the disease.

Bacterial taxonomic composition of tissues and feces associated with healthy and CRC samples. The graphs show the proportion of bacterial families (a) and genera (b) as inferred by PCR amplification and pyrosequencing of the 16S rRNA
Current sequence length of pyrosequencing reads makes taxonomic assignment at the genus level very reliable, but it is not sufficient for accurate species-level assignment. However, species-level PCoA is independent of taxonomic assignment, and allows a graphic representation where samples occupy a position in a three-dimensional (3D) space, depending on their bacterial composition [25]. When a PCoA was performed with the samples grouped by sample type, over 90 % of the variability in the data could be explained by the first three components of the analysis (Fig. 3). In this robust analysis, all three fecal samples clustered closely to each other, indicating that they may not have an appropriate signal to distinguish healthy from diseased individuals. In addition, they had a similar bacterial composition to biopsies from healthy sites. However, the principal component of the analysis clearly separated healthy tissue samples from polyps and tumors, supporting the presence of specific bacterial consortia that are associated with affected sites and that can serve as bacterial biomarkers of colorectal cancer progression. Thus, species-specific detection methods such as qPCR could represent a promising approach for diagnostic purposes.

Principal coordinates analyses (PCoA) based on the bacterial composition of samples included in the study. a Distribution of samples (H correspond to fecal samples; and biopsies are classified as healthy [S], polyp [P] and tumor [T]). b General distribution of all samples included in the study (F feces, B biopsies), which were classified in three groups (Control, Polyp and Tumor)
Mucosal and fecal microbiota composition (Archaea and Bacteria) by qPCR
We observed differences in microbiota composition between patients suffering CRC (n = 15) and those who had normal colon (n = 9). From the four Archaea groups analysed, Methanobacteriales was the unique group detected in both feces and biopsies that were analysed (Tables 2, 3) and we included Methanobrevibacterium and M. smithii as major representative genus and species in human colon. Higher levels of Bifidobacterium spp. (P = 0.069) and lower levels of Methanobacteriales (P = 0.040) and Methanobrevibacterium (P = 0.030) were present in fecal samples from healthy subjects (n = 9) compared to the CRC group (polyp and tumor, n = 15). Higher presence of Fusobacterium nucleatum was observed in fecal samples from CRC compared to controls (9/15, 60 vs. 2/9, 22.2 % and χ 2 test P = 0.07, respectively). Interestingly, we also found higher levels and higher abundance of Enterobacteriaceae in mucosal tissue samples from CRC groups than in controls (P = 0.034 and prevalences 7/15, 46.6 % vs. 0/5, 0 % and χ 2 test P = 0.05, respectively). Faecalibacterium prausnitzii presence was also lower in the CRC group compared to controls (2/15, 13.3 vs. 2/5, 40 % and χ 2 test P = 0.06); however the presence of Akkermansia muciniphila was higher in mucosal samples from CRC than in controls (5/15, 33.3 vs. 0/5, 0 %, and χ 2 test P = 0.136). We also observed a higher presence of Blautia coccoides in CRC than in control samples (13/15, 86.7 vs. 2/5, 40 % and χ 2 test P = 0.036).
When CRC samples are divided in two different disease phases, tubular adenoma (polyp group) and adenocarcinoma (tumor group), we also observed microbiota composition differences in both feces and mucosal samples (Tables 2, 3). Lower, although not statistically significant levels of Bifidobacterium spp. (P = 0.081) and higher levels of Lactobacillus (P = 0.064) were detected in fecal samples from controls (n = 9) compared to those observed in the tumor group (n = 7). Significantly higher levels of Enterobacteriaceae family were found in mucosal samples from tumor compared to polyp groups (P = 0.035).
Significantly higher levels of fecal methanobacteriales were observed when comparing the control group and the tumor group (P = 0.0033), but no differences between controls and adenoma were observed (colonic polyp) (P = 0.48). Additionally, no significant differences between tumor and polyp samples were found (P = 0.189). Methanobrevibacterium was also significantly higher in fecal samples from the tumor group than in controls (P = 0.003); however no differences were found in M. smithii (P = 0.136). We observed a correlation between the CRC process development and fecal levels of methanobacteriales (R = 0.537, P = 0.007), as well as with the levels of Methanobrevibacterium genus (R = 0.574, P = 0.03).
Mucosal and fecal microbiota composition in health and disease
Differences in bacterial diversity were observed in mucosal tissue samples (healthy and CRC tissue samples) from the same individual. We observed different microbial profiles between mucosal samples with polyp and adenocarcinoma compared to their corresponding healthy tissue section. Lower abundances of Staphylococcus and Bacillus and higher abundances of Escherichia-Shigella and Prevotella were observed in mucosal samples from polyps compared to the other tissues, suggesting the potential of those bacterial groups for the diagnosis of CRC development. The abundance of Streptococcus together with Escherichia-Shigella were remarkable in polyps and tumors compared to their corresponding healthy tissue and to the control group. The above results are very promising in the search of disease biomarkers, but a non-invasive sample like fecal material would be more appropriate for developing a diagnostic protocol. Although the PCoA performed showed that fecal samples are not as informative as biopsies, several microorganisms were found to differ between healthy and diseased samples for which fecal material as well as healthy and diseased tissue samples were available for the same individuals.
The adenocarcinomas were classified according to TNM system (Table 1). Although the number of samples is not sufficient to make a robust statistical comparison, we analyzed the impact of tumor severity (T2 and T3) on microbiota composition in order to find a potential biomarker in non-invasive samples. Greater severity of the tumor was related with higher abundance of Parabacteroides (0.9 % in T2 vs. 2.3 % in T3, T test P = 0.03) and non-significant lower abundances of Prevotella (20.2 % in T2 vs. 7.4 % in T3, T test P = 0.802) and Faecalibacterium spp (8.7 % in T2 vs. 6.1 % in T3, T test P = 0.203) in fecal samples, suggesting those bacterial groups as potential biomarkers of the developmental CRC process. We also checked the impact of tumor severity in mucosal samples, and observed lower frequency of Staphylococcus in advanced tumors (31.5 % in T2 vs. 3.2 % in T3, T test P = 0.049). Higher abundance of Staphylococcus was detected in T3 mucosal biopsies from healthy tissues compared to adenocarcinoma tissue (14.9 % in healthy tissue vs. 3.1 % in tumor tissue, T test P = 0.05). Higher abundance of Staphylococcus was related to lower malignancy and was also associated with healthy tissues (49.46 % in T2 healthy tissue; 31.46 % in T2 adenocarcinoma tissue; 14.86 in T3 healthy tissue and 3.12 % in T3 adenocarcinoma tissue). Furthermore, in T3 status, we also found a lower abundance of Streptococcus in healthy than in adenocarcinoma tissue (1.67 % in healthy tissue vs. 8.8 % in tumor tissue, T test P = 0.09). This trend was also observed in tumor tissues between T2 and T3 groups (1.5 % in T2 vs. 8.8 % in T3, T test P = 0.13). Although no significant differences were found in the Escherichia-Shigella group, we observed an interesting trend, where increased abundance of this group was related to tumor status (T2 vs. T3), and also with both mucosal tissue samples (0.22 % in T2 healthy tissue; 0.97 % in T2 adenocarcinoma tissue; 2.85 % in T3 healthy tissue and 13.22 % in T3 adenocarcinoma tissue).
Discussion
Although the etiology of colorectal cancer (CRC) is multifactorial and complex, there is increasing experimental evidence that gut microbiota and their metabolism are linked to CRC development [28]. Whether the contribution to CRC process is generated through intestinal microbial dysbiosis or by specific pathogens is still under discussion. However, host–microbe interactions are mediated by different processes that may include the stimulation of immune system, metabolism and gene expression [29]. Previous studies using both culture-dependent and culture-independent techniques have helped to elucidate the association of one or more microbial species with CRC. Although there is neither clear definition nor right distinction between healthy or normal microbiomes, recent efforts have begun to identify specific perturbations that are indicative of some cancer disease states [28, 30], and some specific gut bacteria have been associated with the pathogenesis of CRC [4, 13, 31]. Similar microbial dysbioses have been described in inflamed colonic mucosa from patients with Inflammatory Bowel Disease (IBD), which has been considered to increase the risk of CRC, compared to control ones [32]. Recently, it has been described that intestinal microbiota also change after surgery for CRC [33].
In this study, we compared the microbial population in mucosa samples of CRC patients presenting polyps or colonic tumors vs. healthy controls. The higher bacterial diversity detected at diseased sites is contrary to the general trend of higher diversity observed in healthy human tissues in inflammatory or infectious diseases compared to diseased sites. This has been interpreted as enrichment at diseased sites of a reduced number of microorganisms that thrive at the specific conditions of the new niche, at the expense of many other microbial species that would be more sensitive to environmental changes. In addition, the lower bacterial diversity detected in tumors could be due to a hampered immune response in the diseased tissue. However, the observed higher microbial diversity in tumor-associated sites compared to normal tissue has also been reported previously [34]. This could be the consequence of the intense irrigation of tumors and polyps, which probably enriches the tissue in nutrients, supporting a higher microbial diversity.
It has been demonstrated that Clostridium leptum and Clostridium coccoides (reclassified as Blautia coccoides) subgroups were specific to CRC and polyposis [15]. We also observed a higher presence of Blautia coccoides detected by qPCR in both fecal and mucosal samples from CRC groups when compared to the control group, and the presence was higher in samples belonging to the adenocarcinoma group, suggesting that these organisms could be interpreted as biomarkers of disease development.
Similar results were obtained by pyrosequencing, where specific groups such as Blautia and Prevotella were virtually absent in healthy tissues, but were present in similar proportion in polyps and tumors, and could therefore be used as potential disease biomarkers. In line with that observation, we found a higher presence of Prevotella in fecal samples from CRC than in healthy samples (29.2 % vs. 14.2 %, respectively; T test P = 0.09). In addition, the number of sequences of Prevotella increased significantly from healthy controls (0.08 % from total) to polyp (2.93 % from total) and tumor (3.06 % from total) groups. By qPCR, we observed a higher presence of the Bacteroides-Prevotella group in mucosal CRC samples (polyp and tumor) than in healthy controls (11/15, 73.3 vs. 2/5, 40 % respectively, χ 2 test P > 0.05). In addition, the number of sequences of Bacteroides increased significantly from healthy controls (2.28 % from total) to polyps (2.93 % from total) and tumors (4.97 % from total). Thus, our results are in agreement with previous reports showing that members of the genus Bacteroides have higher colonization rates in CRC patients [13].
Furthermore, an increased presence of Enterococcus spp. in CRC fecal samples has been reported [31, 35, 36]. Our data showed that Enterococcaceae family was present in all samples, but its proportion was higher in the polyp group (1.5 % from total) than in healthy controls (<0.1 % from total) and adenocarcinoma (0.44 % from total). These data may suggest that higher presence of this group could be a biomarker to detect polyps and a risk factor of CRC.
Several studies have reported the role of mucin-degrading bacteria such as Akkermansia muciniphila (Verrucomicrobia group) in the pathogenesis of inflammatory diseases [37]. We found a higher (although not statistically significant) presence of A. muciniphila in CRC groups than in healthy subjects in mucosal biopsies (5/15, 33.3 vs. 0/5, 0 %, respectively; χ 2 test P > 0.05). In agreement with our data, a recent study showed that Akkermansia muciniphila was about fourfold higher in CRC (P < 0.01) than in control patients [38].
Metagenomic analyses indicate that Fusobacterium species are linked to CRC [17, 28, 39], but their potential role in the disease progression remains unclear. Our observations further confirm that trend, as we observed a significantly higher presence of Fusobacterium nucleatum in mucosal samples from the CRC group (4/15, 26.6 %), compared to the healthy group (0/5, 0 %). In line with our results, increased frequencies of Fusobacterium species were also reported in CRC tumor tissues compared to controls [17, 28, 40]. Recent studies reported higher presence of Fusobacterium spp. in human colonic adenomas (polyps) and also, in stool samples from adenoma and tumor carcinoma patients compared to healthy subjects [28, 40, 41]. Within the CRC group, we also observed differences in the presence of Fusobacterium nucleatum between adenoma (polyp) and tumor groups. Higher presence of F. nucleatum was observed in tumors than in polyp groups in both fecal (6/7, 85.7 vs. 3/8, 37.5 %, respectively and χ 2 test P = 0.05) and mucosal samples (2/7, 28.6 vs. 2/8, 25.0 %, respectively and χ 2 test P > 0.05). By pyrosequencing, we also confirmed the higher presence of Fusobacterium in samples from CRC compared to controls in both feces and mucosal biopsies. Thus, a higher presence of Fusobacterium would be representative of high risk of CRC, and also a potential biomarker of carcinogenesis development. We also found that higher levels of Fusobacterium were related to lower levels of Faecalibacterium prausnitzii. Although we did not observe significant differences in the levels of F. prausnitzii in feces and mucosal biopsies, we observed a tendency suggesting a higher presence of this bacterium in healthy mucosal samples compared to those observed in CRC groups (2/5, 40 vs. 2/15, 13.3 % respectively, and χ 2 test P = 0.19). It has been reported that F. prausnitzii levels decreased significantly in CRC patients compared to healthy subjects [31, 35]. Thus, our data, together with previous studies, suggest a key role of the butyrate-producing bacteria F. prausnitzii as a healthy biomarker in the prevention of CRC. We also observed higher levels of Bifidobacterium spp. in fecal samples from the healthy group compared to the CRC groups. Bifidobacterium species have been suggested as key biomarkers of a healthy gut, and are hypothesized to affect host–microbe interaction, immune modulation and inflammatory response [42]. It has been reported that lower levels of Bifidobacterium spp. have been related to inflammatory related-diseases such as allergy, metabolic disorders and obesity [26, 43, 44]. Chronic inflammation is thought to be the leading cause of CRC, but the mechanisms involved in this process are still poorly understood. It has been suggested that there is an over-reaction of the immune system toward antigens of the gut microbiota, leading to chronic inflammation [45]. An imbalanced gut microbiota composition may trigger a low-grade inflammatory state, or ‘metabolic endotoxaemia’, by rendering the host liable to systemic exposure to the lipopolysaccharide (LPS), a large glycolipid derived from the outer membrane of Gram-negative bacteria, mainly of the Enterobacteriaceae family [46]. In consonance with these data, we observed that the levels of Bifidobacterium spp. were negatively correlated to enterobacterial levels, and we observed higher levels of Bifidobacteria in healthy samples, indicating that this group would be an interesting health biomarker and suggesting their potential use as probiotics to modulate the microbiota and to impact on the immune system and inflammation. The potential effects of probiotics on cancer are well documented [47]. It has been suggested that beneficial bacteria present in the gut microbiota and/or probiotics such as Lactobacillus and Bifidobacterium species have the potential to inhibit the development and progression of the intestinal cell proliferation [48, 49]. Further experimental models and clinical trials are needed to understand the role of probiotics on colon cancer prevention and development, as well as to investigate the exact mechanisms involved in the disease.
The characterization of methanogens in the human colon and its role with health and disease remains incomplete. Methanobrevibacter smithii is the predominant methanogen in the human colon [50, 51]. There is an increasing interest in the archaea populations in the gut and their role in human health. Archaea spp. have been suggested as environmental factors in some diseases, such as obesity [52, 53] and pneumatosis cystoides intestinalis [54]. We observed higher levels of Methanobacteriales in samples from CRC group (polyps and tumors) compared to healthy ones. Significantly higher levels of Methanobacteriales were found in adenocarcinoma than in tubular adenoma (polyp). Our data are in agreement with previous studies where it has been shown that higher levels of breath methane have been reported in patients with ulcerative colitis, intestinal polyposis and tumors [55, 56] compared to healthy patients. However, another study [19] reported higher levels of methanogens (range 45–50 %) in CRC, polyps, and also in the healthy control group. Thus, further studies are needed to understand the role of methanogens on CRC.
In summary, we have identified a different level of diversity in healthy and CRC samples, as well a dysbiosis signature of the gut microbiota in two stages of CRC, tubular adenoma and adenocarcinoma, using a case–control study. Mucosal samples represented the most distinctive microbiota profile allowing the discrimination between two stages of the CRC progress. There are exclusive microbial families and genera present in each group (Fig. 4), suggesting the potential existence of disease biomarkers. Our study showed that observed abundance and detection rate of species belonging to the Fusobacterium, Bacteroides, and Methanobacteriales may provide a potential marker for early detection of CRC. Further data on the relationship between these diverse organisms and CRC etiology may help monitor an individual’s microbiota for early detection of CRC, as well as to design preventive strategies based on the administration of probiotic strains.

Venn diagrams at the family level (a) and genus taxonomic levels (b). Data indicate the number of shared and unique taxa in the microbiota present in mucosal tissue samples according to the type of biopsy (Control, Polyp and Tumor)
References
Cunningham D, Atki W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, Starling N. Colorectal cancer. Lancet. 2010;375:1030–47.
Regula J, Rupinski M, Kraszewska E, Polkowski M, Pachlewski J, Orlowska J, et al. Colonoscopy in colorectal cancer screening for detection of advanced neoplasia. N Engl J Med. 2006;355:1863–72.
Bamia C, Lagiou P, Buckland G, Grioni S, Agnoli C, Taylor AJ, et al. Mediterranean diet and colorectal cancer risk: results form a European cohort. Eur J Epidemiol. 2013;28:317–28.
Sobhani I, Amiot A, Le Baleur Y, Levy M, Auriault ML, Van Nhieu JT, et al. Microbial dysbiosis and colon carcinogenesis: could colon cancer be considered a bacteria-related disease? Therap Adv Gastroenterol. 2013;6:215–29.
Gaboriau-Routhiau V, Lécuyer E, Cerf-Bensussan N. Role of microbiota in postnatal maturation of intestinal T-cell responses. Curr Opin Gastroenterol. 2011;27(6):502–8.
O’Keefe SJ, Chung D, Mahmoud N, Sepulveda AR, Manafe M, et al. Why do African Americans get more colon cancer than Native Africans? J Nutr. 2007;137:S75–82.
Man SM, Kaakoush NO, Mitchell HM. The role of bacteria and pattern-recognition receptor’s in Crohn’s disease. Nat Rev Gastroenterol Hepatol. 2011;8:152–68.
Candela M, Guidotti M, Fabbri A, Brigidi P, Franceschi C, Fiorentini C. Human intestinal microbiota: cross talk with the host and its potential role in colorectal cancer. Crit Rev Micro. 2011;37:1–14.
Giardiello FM, Brensinger JD, Petersen GM. AGA technical review on hereditary colorectal cancer and genetic testing. Gastroenterology. 2001;21:198–213.
Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482(7384):179–85.
Marchesi JR, Dutilh BE, Hall N, Peters WHM, Roelofs R, et al. Towards the human colorectal cancer microbiome. PLoS ONE. 2011;6(5):e20447.
Hu B, Elinav E, Huber S, Strowig T, Hao L, Hafemann A, et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. PNAS. 2013;110(24):9682–7.
Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, Corthier G, Tran Van Nhieu J, Furet JP. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS ONE. 2011;6(1):e16393.
Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS ONE. 2012;7(6):e39743.
Scanlan PD, Shanahan F, Clune Y, Collins JK, O’Sullivan GC, O’Riordan M, et al. Culture-independent analysis of the gut microbiota in colorectal cancer and polyposis. Environ Microbiol. 2008;10:789–98.
Parkin D. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118:3030–44.
Castellarin M, Warren R, Freeman J, Dreolini L, Krzywinski M, Strauss J, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22:299–306.
Derrien M, Vaughan EE, Plugge CM, de Vos WM. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol. 2004;54(Pt 5):1469–76.
Scanlan PD, Shanahan F, Marchesi JR. Human methanogen diversity and incidence in healthy and diseased colonic groups using mcrA gene analysis. BMC Microbiol. 2008;20(8):79.
Azcárate-Peril MA, Sikes M, Bruno-Bárcena JM. The intestinal microbiota, gastrointestinal environment and colorectal cancer: a putative role for probiotics in prevention of colorectal cancer? Am J Physiol Gastrointest Liver Physiol. 2011;301(3):G401–24.
Cabrera-Rubio R, Garcia-Núñez M, Setó L, Antó JM, Moya A, Monsó E, Mira A. Microbiome diversity in the bronchial tracts of patients with chronic obstructive pulmonary disease. J Clin Microbiol. 2012;50:3562–8.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–41.
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, et al. The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–5.
Yarza P, Richter M, Peplies J, Euzeby J, Amann R, Schleifer K, et al. The all-species living tree PROJECT: a 16S rRNA-based phylogenetic tree of all sequenced type strains. Syst Appl Microbiol. 2008;31:241–50.
Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71(12):8228–35.
Collado MC, Isolauri E, Laitinen K, Salminen S. Distinct composition of gut microbiota during pregnancy in overweight and normal-weight women. Am J Clin Nutr. 2008;88:894–9.
Yu Y, Lee C, Kim J, Hwang S. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol Bioeng. 2005;89(6):670–9.
Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14(2):207–15.
Turner ND, Ritchie LE, Bresalier RS, Chapkin RS. The microbiome and colorectal neoplasia: environmental modifiers of dysbiosis. Curr Gastroenterol Rep. 2013;15(9):346.
Vipperia K, O’Keefe SJ. The microbiota and its metabolites in colonic mucosal health and cancer risk. Nutr Clin Practice. 2013;27:624–35.
Wu N, Yang X, Zhang R, Li J, Xiao X, Hu Y, et al. Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb Ecol. 2013;66(2):462–70.
Bibiloni R, Mangold M, Madsen KL, Fedorak RN, Tannock GW. The bacteriology of biopsies differs between newly diagnosed, untreated, Crohn’s disease and ulcerative colitis patients. J Med Microbiol. 2006;55:1141–9.
Ohigashi S, Sudo K, Kobayashi D, Takahashi T, Nomoto K, Onodera H. Significant changes in the intestinal environment after surgery in patients with colorectal cancer. J Gastrointest Surg. 2013;17(9):1657–64.
Geng J, Fan H, Tang X, Zhai H, Zhang Z. Diversified pattern of the human colorectal cancer microbiome. Gut Pathog. 2013;5(1):2.
Balamurugan R, Rajendiran E, George S, Samuel GV, Ramakrishna BS. Real-time polymerase chain reaction quantification of specific butyrate-producing bacteria, Desulfovibrio and Enterococcus faecalis in the feces of patients with colorectal cancer. J Gastroenterol Hepatol. 2008;23(8 Pt 1):1298–303.
Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang X, Jia W, Cai S, Zhao L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012;6(2):320–9.
Shih DQ, Targan SR. Immunopathogenesis of inflammatory bowel disease. World J Gastroenterol. 2008;14(3):390–400.
Weir TL, Manter DK, Sheflin AM, Barnett BA, Heuberger AL, Ryan EP. Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS ONE. 2013;8(8):e70803.
Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2011;22(2):299–306.
Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2011;22(2):292–8.
McCoy AN, Araújo-Pérez F, Azcárate-Peril A, Yeh JJ, Sandler RS, Keku TO. Fusobacterium is associated with colorectal adenomas. PLoS ONE. 2013;8(1):e53653.
Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57(6):1470–81.
Kalliomäki M, Kirjavainen P, Eerola E, Kero P, Salminen S, Isolauri E. Distinct patterns of neonatal gut microflora in infants developing or not developing atopy. J Allergy Clin Immunol. 2001;107:129–34.
Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–7.
Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annu Rev Immunol. 2002;20:495–549.
Cani PD, Osto M, Geurts L, Everard A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes. 2012;3(4):279–88.
Lyra A, Lahtinen S, Ouwehand AC. Gastrointestinal benefits of probiotics: clinical evidence. In: Salminen S, von Wright A, Lahtinen S, Ouwehand A, editors. Lactic acid bacteria: microbiological and functional aspects. 4th ed. Boca Raton: CRC Press; 2012. p. 509–23.
Geier MS, Butler RN, Howarth GS. Probiotics, prebiotics and synbiotics: role in chemoprevention for colorectal cancer? Cancer Biol Ther. 2006;5(10):1265–9.
Collado MC, Isolauri E, Salminen S, Sanz Y. The impact of probiotic on gut health. Curr Drug Metab. 2009;10(1):68–78.
Abell GCJ, Conlon MA, McOrist AL. Methanogenic archaea in adult human faecal samples are inversely related to butyrate concentration. Microb Ecol Health Dis. 2006;18:154–60.
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–8.
Mathur R, Amichai M, Chua KS, Mirocha J, Barlow GM, Pimentel M. Methane and hydrogen positivity on breath test is associated with greater body mass index and body fat. J Clin Endocrinol Metab. 2013;98(4):E698–702.
Million M, Angelakis E, Maraninchi M, Henry M, Giorgi R, Valero R, et al. Correlation between body mass index and gut concentrations of Lactobacillus reuteri, Bifidobacterium animalis, Methanobrevibacter smithii and Escherichia coli. Int J Obes (Lond). 2013;37(11):1460–6.
Florin TH. Alkyl halides, super hydrogen production and the pathogenesis of pneumatosis cystoides coli. Gut. 1997;41:778–84.
Pique JM, Pallares M, Cuso E, Vilar-Bonet J, Gassull MA. Methane production and colon cancer. Gastroenterology. 1984;87:601–5.
Holma R, Osterlund P, Sairanen U, Blom M, Rautio M, Korpela R. Colonic methanogenesis in vivo and in vitro and fecal pH after resection of colorectal cancer and in healthy intact colon. Int J Colorectal Dis. 2012;27(2):171–8.
Acknowledgments
EntreChem S.L. also acknowledges funding from FICYT (project IE-09-314).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
L. Mira-Pascual, R. Cabrera-Rubio and S. Ocon contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mira-Pascual, L., Cabrera-Rubio, R., Ocon, S. et al. Microbial mucosal colonic shifts associated with the development of colorectal cancer reveal the presence of different bacterial and archaeal biomarkers. J Gastroenterol 50, 167–179 (2015). https://doi.org/10.1007/s00535-014-0963-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-014-0963-x