
Abstract
The interpretation of colonic biopsies related to inflammatory conditions can be challenging because the colorectal mucosa has a limited repertoire of morphologic responses to various injurious agents. Only few processes have specific diagnostic features, and many of the various histological patterns reflect severity and duration of the disease. Importantly the correlation with endoscopic and clinical information is often cardinal to arrive at a specific diagnosis in many cases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The increasing use of colonoscopy and biopsy to investigate patients presenting with gastrointestinal symptoms has led to a better understanding and appreciation of the histopathologic changes underlying inflammatory conditions of the large bowel. In this review, we present the spectrum of non-idiopathic inflammatory bowel disease conditions of the colon, excluding drug-induced colitis. Interpretation of colonoscopic biopsies in the diagnosis of these various entities will be emphasized and differential diagnosis discussed, including the differentiation from idiopathic inflammatory bowel disease (IBD) where relevant.
Practically, a structured approach should be adopted to avoid missing important diagnostic clues. Each compartment should be assessed, including epithelium, lamina propria, muscularis mucosae, and submucosa, if present.
Infectious colitis
Acute infectious proctocolitis refers to an inflammatory condition due to a potentially identifiable infectious cause. The disease is usually self-limiting, most resolving within 2–4 weeks or less, without residual inflammation or recurrent symptoms. Since a proportion of cases return a negative culture, the term “acute infectious-type colitis” or “acute self-limiting colitis” is commonly used to refer to patients who present with a transient diarrheal illness, with biopsy findings similar to those with an identifiable infectious etiology, but in whom no pathogen is identified [1].
Among the viral causes of diarrhea, norovirus and rotavirus are the most common in the USA, accounting for 30–40% of diarrheal illnesses [2]. Common bacterial causes of diarrhea include Campylobacter, Shigella, non-typhoidal Salmonella species, and Escherichia coli acquired as a result of consumption of undercooked meat or poultry, fecally contaminated water, and in travelers [2–4]. Yersinia is a relatively common cause of bacterial enterocolitis in Northern and Western Europe and increasing numbers of cases are being recognized in the USA and Australia [3]. Aeromonas species has been increasingly recognized as a cause of gastroenteritis both in pediatric and adult populations [3]. Finally, clostridial organisms are well-known causes of antibiotic associated and nosocomial infections [2, 3]. Among parasitic infections, the intestinal protozoan Entamoeba histolytica is one of the more important causes of infectious colitis in Western countries, often but not invariably seen in travelers to endemic regions [2, 4]. Fungal infections are uncommon in industrialized nations and are usually encountered as opportunistic infections in immunologically compromised hosts [5].
The presentation typically includes acute onset diarrhea, which may be bloody, with abdominal cramps and variable systemic symptoms including fever [6, 7]. Colonoscopic findings include mucosal granularity, erosions, ulcers, bleeding, and friability [6, 7]. Pseudomembranes and mass lesions may occasionally be seen [3, 7]. Most common bacterial and viral agents produce self-limiting illness; however, some may require appropriate supportive antimicrobial treatment [2–4].
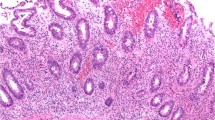
Most etiologies produce a similar pattern referred to as an acute infectious-type (self-limiting) colitis pattern (ASLC) [1]. The exact histologic picture, however, will depend on the time of the biopsy relative to the onset of the infection. The classic appearance is seen when the biopsies are taken within the first week of illness and is characterized by preserved mucosal architecture, lamina propria edema, and neutrophils within the superficial lamina propria (Fig. 1). The neutrophilic infiltrate may be patchy and found adjacent to ectatic capillaries, with marginating neutrophils; or invading the mucosal surface, sometimes with intercryptal epithelial tufts, and cryptitis, which when present is typically superficial. Crypt abscesses may be seen but are generally not common. Accompanying epithelial injury variably includes mucin depletion, flattening of epithelial cells, and withering crypts.

Acute self-limiting colitis (ASLC) pattern. There is preservation of the crypt architecture with mucin depletion and reactive epithelial changes (a H&E). The lamina propria contains increased numbers of inflammatory cells; however, at higher magnification neutrophils (arrowhead) are seen superficially with superficial cryptitis characterized by neutrophil infiltration of crypt epithelium (arrow) without crypt abscesses (b H&E)
Early in the course, edema of the lamina propria with clusters of neutrophils and ectatic capillaries are the sole findings. Two to 4 weeks into the course of the disease, there may be appreciable increase in mononuclear inflammatory cells within the lamina propria with occasional basal lymphoid aggregates. As resolution occurs, there is remission of neutrophilic infiltrate but the increase in mononuclear cells in the lamina propria may persist with or without basal lymphoid aggregates, giving an appearance that may suggest IBD, particularly Crohn’s disease [1].
Features favoring an infectious colitis include predominance of neutrophils over mononuclear cells or an exclusively neutrophilic infiltrate (often superficially located) in the lamina propria, scattered superficial cryptitis, and preservation of crypt architecture. On the other hand, features favoring IBD include numerous crypt abscesses, increased transmucosal lamina propria lymphoplasmacytic infiltrate with basal plasmacytosis with or without basal lymphoid aggregates, crypt architectural distortion, villiform transformation, basal giant cells, and epithelioid granulomas [6, 8].
The features suggestive of ASLC are best seen within the first week of symptomatology. Later, the diagnosis of ASLC is mainly one of inference based on the absence of characteristic features of IBD [1, 8]. Importantly, if severe and/or prolonged, infections with Campylobacter, Shigella, and Aeromonas may produce architectural distortion mimicking IBD [3]. In such cases clinical and endoscopic follow-up will be cardinal to the diagnosis since full resolution of changes occurs within 2–3 weeks in most infectious colitis and virtually always within 3 months [6].
Epithelial degeneration with withering crypts may raise the differential diagnosis of ischemic colitis; however, true ischemia will be associated with other features (e.g., lamina propria hyalinization, fibrin thrombi, hemosiderin) whilst the presence of other features of an ASLC pattern points to an infectious colitis.
If intercryptal epithelial syncytial tufts are noted, it is important to inquire about a history of antibiotic use, and if present additional step sections are advised to exclude a type I lesion of pseudomembranous colitis (see later).
Finally, certain drugs, strong purgatives such as hypertonic enemas, and laxative abuse may sometimes cause a similar picture to acute infectious-type colitis (self-limiting) colitis [9]. Knowledge of the clinical presentation is therefore important.
Infections producing specific histologic appearances
Enterohemorrhagic E. coli (EHEC)
The most common strain of EHEC is the O157:H7 often linked to outbreaks of infectious diarrhea related to consumption of undercooked hamburger patties [4]. Transmission may also occur through contaminated water, unpasteurized milk, and person-to-person contact [10]. The pathogenicity is related to the production of adherence factors mediating adherence to the epithelial surface and the elaboration of two Shiga-like toxins; however, the organism does not invade the mucosa [2].
Following a short incubation of 1–4 days, usually bloody diarrhea develops associated with severe abdominal cramps and mild or no fever [3, 11]. Fecal leukocytes are noted in up to one-third of cases [3]. Hemolytic uremic syndrome or thrombocytopenic purpura may develop particularly in children or the elderly [12, 13]. Endoscopically, mucosal edema, erosions, ulcers, and hemorrhage can be seen. The right colon is usually more severely affected [3]. The illness usually lasts less than 10 days [3, 12].
A spectrum of histologic changes from acute infectious-type colitis pattern to ischemic pattern or with overlapping ischemic and infectious features can be seen. Intramural fibrin deposition and fibrin thrombi in mucosal and submucosal capillaries may be observed. Pseudomembranes and transmucosal necrosis with mucosal sloughing and ulceration may occur as well [3, 14].
The differential diagnosis includes Clostridium difficile colitis and ischemic colitis, which show overlapping features and may occasionally be difficult or impossible to exclude [12, 14].
C. difficile-associated colitis and pseudomembranous colitis
Traditional risk factors include recent antibiotic therapy, older patient age, previous gastrointestinal disease or surgery, acid suppression, chemotherapy, and recent hospitalization or stay in a chronic care facility [15]. However, the epidemiology of C. difficile is changing and the infection is now being reported in populations previously not considered to be at risk, such as pregnant women, children, and patients with IBD [16]. Proton pump inhibitor (PPI) therapy may also predispose to community-acquired C. difficile colitis [16].
C. difficile is a spore-forming Gram-positive anaerobe that exerts its pathogenic effects through the production of toxins, the most important being toxin A and toxin B that result in disruption of the cellular cytoskeleton, loosening of intercellular tight junctions, and a profound secretory diarrhea [16]. Although most pathogenic strains produce both toxins, 2–56% of C. difficile-associated infection is attributable to strains of C. difficile producing toxin B alone [17].
The clinical features of C. difficile infection can vary from asymptomatic carriage to fulminant colitis [15]. In symptomatic patients, watery diarrhea is occasionally associated with mucus or blood. Stool frequency may exceed 10 times per day and the stool may have a characteristic foul odor [15]. Lower abdominal cramps, fever, anorexia, malaise, and dehydration may occur in severe cases. On colonoscopy, edema, blurring of the vascular pattern, and thickening and blunting of the haustral folds are observed. Pseudomembranes are seen as discrete creamy-yellow plaques separated by congested mucosa (Fig. 2a) [18]. In severe cases, coalescence of pseudomembranes occurs with the resulting necrotic membranes resembling ischemic colitis [19]. Toxic megacolon and perforation may develop [20].

Pseudomembranous colitis. Characteristic creamy-yellow plaques separated by congested mucosa are seen on colonoscopy (a). Microscopically, characteristic features include disrupted crypts distended by mucin, fibrin, neutrophils, and eosinophils that form the classic inflammatory pseudomembrane of the surface. In addition, pseudosignet-ring cells (arrow) are also seen (b H&E)
Histologically, the earliest lesions (type I lesions) consist of focal intercryptal surface erosion with a luminal spray of nuclear debris, neutrophils, and mucin [21]. The subjacent lamina propria is edematous with clusters of neutrophils and dilated capillaries. Focal cryptitis and a pink subepithelial exudate may be present. Intact surface epithelial cells may be tufting or appear crenated with infiltrating neutrophils. Later lesions demonstrate the classic picture that corresponds to plaques noted endoscopically. Type II lesions consist of a small focus of disrupted crypts distended by mucus, fibrin, neutrophils, and eosinophils (Fig. 2b) [19]. The epithelia of the affected crypts are flattened and progressively lost, initially superficially. The mucin, fibrin, neutrophils, and eosinophils stream out of the dilated crypts and coalesce to form an inflammatory pseudomembrane over the surface with characteristic linear streaming of neutrophils/eosinophils within fibrin strands and mucus. Occasional fibrin thrombi are found in superficial mucosal capillaries. The submucosa is edematous. As the lesions enlarge, the crypt destruction becomes complete (type III lesion) with a layer of fibroinflammatory exudate, mucus, and cell debris covering the residual mucosa [19]. Occasionally, degenerating pseudosignet-ring cells are found but are usually confined within the basement membrane of the crypts without infiltration [22]. The nuclei of the pseudosignet-ring cells are not enlarged, have uniform chromatin and inconspicuous nuclei. These pseudosignet-ring cells are negative for p53 and Ki-67 and positive for E-cadherin [23].
The differential diagnosis of pseudomembranous colitis varies depending on the stage of development. Lesions similar to type I lesions can be seen with acute infectious colitis or mucosal prolapse. It is important to examine the adjacent mucosa for accompanying clues [19]. For example, in solitary rectal ulcer splaying of the muscularis mucosa with fibromuscular obliteration of the lamina propria will be noted. In type II and III lesions, the appearances may overlap with ischemic colitis [24]. Hyalinization of the lamina propria, hemorrhage, withering microcrypts, full-thickness mucosal necrosis, and diffuse microscopic distribution of pseudomembranes favor ischemia [24]. On the other hand, diffuse distribution of punctate pseudomembranes endoscopically is characteristic of C. difficile [24].
Yersinia species-related enterocolitis
The Gram-negative facultative anaerobic coccobacilli Yersinia enterocolitica and Yersinia pseudotuberculosis may cause a bacterial enterocolitis, sometimes mimicking Crohn’s disease and acute appendicitis [25]. Yersinia has been found in fish, shellfish, swine, cattle, milk, ice cream, beef, lamb, poultry, and pork [3, 25]. Undercooked food and contaminated milk are the most common sources of infection and children are affected more often than adults [25]. Cirrhotic and immunosuppressed patients, diabetics, and elderly and malnourished individuals are also at an increased risk [25]. Other predisposing factors include hemochromatosis, acute iron poisoning, and transfusion-dependent blood dyscrasias [25]. Both Y. enterocolitica and Y. pseudotuberculosis may cause colitis; however, Y. pseudotuberculosis more often produces a terminal ileitis and mesenteric adenitis with granulomas and microabscesses [25, 26]. Both bacterial species can be associated with self-limited enterocolitis to potentially fatal infections, particularly in immunosuppressed and patients with iron overload [25]. Common symptoms include fever, diarrhea, nausea, vomiting, and abdominal pain that may last from 1 to 3 weeks [25]. Fecal leukocytes, blood, or mucus may be present in stool specimens [25]. Classically, mesenteric adenitis or ileitis may be indistinguishable from acute appendicitis [26]. Extraintestinal manifestations may develop including a migratory polyarthritis, Reiter syndrome (common in HLA-B27-positive patients), and erythema nodosum [25, 27]. Colonoscopic examination shows round to oval elevations with or without ulceration in the terminal ileum and yellow oval aphthae of the colon [28]. Pathologic changes are centered on the ileocecal region and appendix; these are thickening of the bowel wall with inflammation, congestion, ulceration, and edema of the mucosa including aphthous ulcers [29]. Mesenteric nodes are typically enlarged and may be matted and contain yellowish microabscesses [29].
Characteristic mucosal changes include prominent lymphoid hyperplasia with overlying aphthous ulcers, covered by fibrinopurulent exudate and large numbers of Gram-positive coccobacilli. Cryptitis and architectural distortion may be seen. Epithelioid granulomas with central suppuration and necrosis and prominent surrounding lymphoid cuffing may be seen in the mucosa (and within the bowel wall). Similar granulomas may also be observed in regional lymph nodes [26, 29].
The differential diagnosis includes Crohn’s disease and tuberculosis, the latter being readily excluded by culture, serological studies, and PCR assays [3, 30]. Acid-fast stains and culture results should help to distinguish mycobacterial infection [3]. Crohn’s disease may be very difficult to exclude. Features that favor Crohn’s include cobblestoning of mucosa and creeping fat grossly, and evidence of chronicity [3]. Stellate abscesses or granulomas with central suppuration and lymphoid cuffing are suggestive of Yersinia infection. However, some cases are indistinguishable on histological grounds [3].
Tuberculosis
Tuberculosis of the gastrointestinal tract, whether primary or secondary, usually involves the terminal ileum, cecum, and appendix, while left colon and rectum are less commonly involved [31]. In the USA, gastrointestinal infection is rare and almost always secondary [31]. Three forms are reported: (1) ulcerative, the most common (~60% of cases) with a virulent course; (2) hypertrophic (10%), mimicking Crohn’s disease and characterized by fibrous scarring and mass-like lesions; and (3) ulcerohypertrophic (30%), with thickening of the intestinal wall, ulceration, and formation of an inflammatory mass [32].
The clinical manifestations are nonspecific and include fever, weight loss, diarrhea, hematochezia, and abdominal pain [33]. Symptoms referable to the respiratory system, joints, and lymphadenopathy can be prominent [33]. On colonoscopy, transversely placed and linear ulcers and hypertrophic inflammatory masses (Fig. 3) can be seen [34]. Notably, strictures and mucosal bridges and edematous or patulous ileocecal valve is characteristic [34].

Tuberculosis. Hypertrophic inflammatory masses are seen on colonoscopy
The histologic hallmark of tuberculosis is the presence of compact aggregates of epithelioid cells and Langhans giant cells with central caseation and surrounding lymphoid cuff [32, 33]. Granulomas may be found, within the lamina propria or submucosa of intact mucosa or at the base of ulcerated areas. Healing granulomas are surrounded by a rim of fibrous tissue in lymph nodes, but not in the intestinal wall [32]. Acid-fast stains may demonstrate typical organisms within granulomas. Intact mucosa may show only nonspecific chronic inflammation but there may be architectural distortion and pyloric metaplasia [32]. Early granulomas are usually found within lymphoid tissue [32, 35]. Fissuring ulcers may be seen as well as fibrosis that may efface the mucosal architecture [32].
The differential diagnoses to consider include Crohn’s disease and yersiniosis and awareness of clinical, histopathologic, and microbiologic investigation should secure a correct diagnosis [3, 33, 34].
Intestinal spirochetosis
It can be detected in 2–7% of colorectal biopsies and in up to 54% in the homosexual population [36]. The most common species arguably causing intestinal disease are Brachyspira aalborgi and Brachyspira pilosicoli [37]. The clinical significance is debated and it is believed that in most instances intestinal spirochetosis represents an incidental finding and likely is not responsible for the patient’s symptomatology [36]. Even in symptomatic patients, coinfection with other organisms should be excluded before a therapeutic trial may be undertaken [36, 38]. The most common form of intestinal spirochetosis is the noninvasive, commensal form. Alternatively, an invasive form with invasion of mucosa leading to enterocolitis, hepatitis, or bacteremia can be recognized, particularly in immunocompromised hosts [36].
Histologically, a thickened 2–3 μm basophilic fringe is observed on the epithelial surface, sparing goblet cells (Fig. 4a) [37]. Periodic acid–Schiff (PAS) reagent or Warthin–Starry stain (Fig. 4b) highlights the bacterial colonization [37, 39]. The colonic mucosa is otherwise relatively unremarkable, although mild or focal increase in inflammation with some cryptitis may be seen [38].

Intestinal spirochetosis. Note the thickened basophilic fringe of the epithelial surface due to the spirochete organisms (a H&E). The organisms are highlighted by a Warthin–Starry stain (b)
Amebiasis
Amebiasis is caused by the protozoan Entamoeba histolytica [40]. Infection is acquired through ingesting contaminated water and food but may also occur by direct feco-oral contact or via anal intercourse [40, 41]. High-risk groups for infection include travelers to endemic regions (tropics), pregnant women, debilitated patients (e.g., malnutrition, alcoholics, diabetics, and cancer patients), immunosuppressed and elderly patients [40, 42].
Following infection, the clinical spectrum ranges from asymptomatic infection to amebic dysentery with bloody diarrhea or to extraintestinal manifestation including hepatic abscesses and less commonly systemic spread with abscesses in other organs [40]. Current diagnostic tools include antigen detection in stool and PCR [40]. On colonoscopy, multiple punctate ulcers may be seen with yellow-white exudate and hyperemic borders (Fig. 5a) [43]. The changes are more common in the right colon but may involve the left colon as well [40].

Amebiasis. Ulceration with yellow-white exudate on colonoscopy (a). Microscopically, superficial erosion is seen with numerous amebic trophozoites within surface exudate, some containing phagocytized red cells, a characteristic feature (arrows). Neutrophils can be seen within lamina propria but crypt abscesses are absent (b H&E)
Histologically, several stages of development have been described [44]. Initially a picture of acute infectious-type (self-limiting) colitis is observed. Microulceration covered by basophilic exudate containing organisms ensues (Fig. 5b). The surrounding mucosa is edematous with neutrophilic infiltration of the lamina propria but few crypt abscesses. In the fully developed lesion, tissue necrosis and ulceration extend into the submucosa and is covered by a thick inflammatory exudate separated from underlying viable tissue by a fibrinoid band. This corresponds to the classic flask-shaped ulcer. The intervening mucosa is congested and infiltrated by neutrophils, plasma cells, and eosinophils. Prolonged amebiasis may mimic IBD with ulceration, architectural changes of chronicity and fibrosis [45]. However, it is important to note that amebiasis may coexist and exacerbate preexisting IBD [46]. Ameba should also be differentiated from Balantidium coli, the trophozoites of which are larger and have two nuclei (a sausage/kidney-shaped macronucleus and adjacent spherical micronucleus that may be obscured) and are ciliate [47].
Diagnosis is established when trophozoites are recognized, usually within the exudate and invading mucosa in the fully developed lesion. The protozoa are large round to oval (6–40 μm) PAS-positive organisms with purple foamy cytoplasm and a small pale nucleus [44]. Erythrophagocytosis is a characteristic finding, which may be highlighted by a Heidenhain iron hematoxylin stain [48].
Cytomegalovirus (CMV) infection
Primary, reactivation, and superinfection represent the three forms of CMV infection [49]. Primary infection is commonly subclinical and produces an infectious mononucleosis-like illness [49]. Documented examples of primary colonic CMV infection are rare. CMV colitis commonly develops as part of disseminated disease, most due to reactivation of latent virus in the immunocompromised host (e.g., HIV/AIDS infection or post transplantation) [50]. Superinfection has been shown to occur particularly in the setting of IBD [51, 52].
CMV colitis presents with abdominal pain and diarrhea, which may be watery or bloody [49, 50]. On colonoscopy, ulcers (38%) or colitis (20%) or both (29%) may occur [50]. In addition to biopsies of granulation tissue from ulcer bases where the yield is highest, blind biopsies may be required if clinically suspected as 25% may have normal-appearing mucosa [50]. The sensitivity of left colon biopsies is approximately 77% and increases to 100% when combined with right colon and ileal biopsies [50].
Microscopically, the detection of intranuclear inclusions within macrophages, fibroblasts, smooth muscle, ganglion cells, or endothelial cells is diagnostic (Fig. 6a, b). Inclusions may be also seen in epithelial cells, and tends to be associated with no or mild inflammation [53]. Typically, the infected cells show nuclear and cytoplasmic enlargement with a single dark amphophilic nuclear “owl’s eye” inclusion [54]. However, indistinct smudged hematoxyphilic nuclei may only be seen [54]. The cytoplasm shows granular basophilic inclusions or may be foamy [54]. The mixed inflammatory response may be minimal or marked with necrosis and ulceration [53, 54].

CMV colitis. Endothelial cells showing CMV cytopathic effect characterized by cytomegaly and nucleomegaly with large amphophilic nuclear inclusions (arrow) (a H&E). Immunoperoxidase stain for CMV showing positive nuclear staining (b)
CMV colitis should enter the differential diagnosis if atypical cells are present in granulation tissue of intensely inflamed and ulcerated biopsies [54]. A careful search for CMV inclusions in macrophages, endothelial cells, granulation tissue, and epithelial cells should be undertaken, and even if negative immunohistochemical staining should be ordered. The histological features in severe infections may also resemble ischemia because of occlusive vasculitis or vasculopathy secondary to infected endothelial cells [54].
Microscopic colitis (MC)
MC is a clinical designation used to describe patients with chronic watery diarrhea and essentially normal endoscopy but with histologic inflammation of the colonic mucosa [55]. It essentially encompasses two histopathologic entities, namely collagenous colitis (CC) and lymphocytic colitis (LC). These conditions, despite similar clinical presentation and some overlapping histopathologic features, are distinctive in many regards [56–58]. Whether they represent separate but related entities or different ends of the spectrum of a single disorder has been debated and examples of cases transitioning from one to another have been reported [59]. The patchiness of subepithelial collagen thickening of CC due to suboptimal sampling may account for the variation of diagnoses in the same patient.
MC is usually diagnosed in late middle age and elderly with a peak incidence in the sixth to seventh decade [59]. Both conditions are uncommon in children [60] and Asia [61]. Female predilection is reported in CC but inconsistently for LC [59]. Both CC and LC present with chronic or recurrent watery diarrhea. Abdominal pain as well as weight loss may occur [59]. The natural history of both conditions is highly variable ranging from chronic relapsing symptomatology to spontaneous remission (more common in LC) [59]. Rare cases of colonic perforation or diarrhea refractory to medical therapy requiring colectomy or diversion of fecal stream have been reported [62–64].
Approximately 40% of patients may have associated autoimmune disease, the most common being celiac disease, rheumatoid arthritis, thyroid disorders, and diabetes [56, 58, 59]. Celiac disease patients may have up to a 70-fold increase in the risk for MC compared to the general population [65]. The association of gluten sensitivity with LC is stronger than with CC [66]. Given this association, exclusion of celiac disease in patients with MC and vice versa should be considered, particularly in cases that remain symptomatic despite treatment [65–67]. MC may also be associated with intraepithelial lymphocytosis of the terminal ileum and primary ileal villous atrophy [68, 69]. Thickening of the subepithelial collagen band in the terminal ileum may also be seen with CC [70, 71]. CC has been described in association with collagenous gastritis in adults [72]. Similarly, gastric intraepithelial lymphocytosis and lymphocytic gastritis may be seen in association with LC [73].
MC has also been associated with medication use and a proportion of cases fulfill the criteria for a drug-induced colitis [74]. Associated medications include NSAIDs, PPIs (esomeprazole, omeprazole, lansoprazole), ranitidine, cyclo-3-fort, SSRIs, beta-blockers, statins, bisphosphonates, ticlopidine, flutamide, acarbose, and isoretinoin [75–77].
Both Crohn’s disease and ulcerative colitis have been seen in association with MC, either preceding or developing after a diagnosis of lymphocytic or collagenous colitis; however, a pathogenic link has not been established [78].
The etiology of MC is not entirely clear. Reports of familial clustering of MC [79], associations with certain HLA haplotypes (HLA-DQ2 or DQ1,3) [80], and polymorphisms in TNF-α gene [81] suggest a possible underlying genetic component. The strong association with autoimmune conditions suggests an underlying immune dysregulation. An abnormal response to a luminal or environmental agent is suggested by the observation of improvement following institution of an elemental diet or diversion of the fecal stream [82]. Potential luminal agents that have been proposed include bacterial toxins, medications, and bile salts [59]. It is possible that MC may represent an abnormal immunological response to a luminal or environmental agent in a genetically susceptible host. The mechanism for diarrhea is incompletely understood. Some studies have found active secretion of sodium and chloride and downregulation of tight-junction molecules leading to increased permeability [83]. The role of the thickened subepithelial collagen table is controversial with suggestion that it may act as a diffusion barrier [83]. However, the severity of the diarrhea appears to correlate with the degree of inflammation rather that the thickness of the subepithelial collagen table [84].
At colonoscopy, the mucosa is typically unremarkable but minimal changes including mild edema or an opalescent appearance, mild erythema, or diminished vascular pattern have been reported [85]. Over the years, there have been an increasing number of reports of colonoscopic abnormalities associated with CC, calling into question its so-called microscopic nature. These include diminished vascular pattern, pruning of mucosal vasculature or crowded dilated tortuous capillaries; red spots, nodularity or textural abnormalities (with or without chromoendoscopy); hemorrhagic lacerations or linear tears more commonly in ascending and transverse colon (either spontaneously or with insufflation) and fine linear cicatricial lines or thick scar like ridges possibly due to healing of mucosal tears/defects [86]. A mosaic or “honeycomb” pattern of the mucosa in the appropriate clinical context of chronic watery diarrhea has been reported by some as a distinguishing feature of CC [86, 87].
Collagenous colitis
Histologically, CC is characterized by increased lamina propria cellularity expanded by lymphocytes and plasma cells as well as increased eosinophils while the crypt architecture is maintained [59]. Limited epithelial injury with loss of the normal columnar shape, mucodepletion, and a flat or syncytial appearance of the surface epithelium may be seen [59]. It is not uncommon for the surface epithelium to become detached and this is often a clue to suspect the diagnosis (Fig. 7) [88]. Increased lymphocytic exocytosis is variably present and of a lesser degree than that observed in LC [89–91]. The characteristic thick collagen band present beneath the surface epithelium does not extend down the sides of the crypts (Fig. 7). The normal subepithelial collagen table is reported to be less than 7 μm and a thickness of greater than 10 μm has classically been required to establish the diagnosis in the proper setting [59, 88]. However, subtle evidence such as the characteristics of the collagen band, i.e., presence of entrapped red cells, inflammatory cells, and capillaries within the band and an irregular jagged inferior aspect can be seen [59, 88]. It should be noted that a thickened subepithelial collagen band, in isolation, is not specific to CC. This can be seen in hyperplastic polyps, adjacent to diverticula, or healing ulcer [92]. Tangential sectioning may result in an artifactually expanded appearance of the normal basement membrane and/or subnuclear cytoplasmic zone of the surface epithelium mimicking a thickened subepithelial collagen table [88, 92]. It should also be remembered that thickening of the subepithelial collagen band may be patchy and show regional variation with more frequent and marked expansion observed on the right colon as compared to distal colon [59]. Therefore, flexible sigmoidoscopy and left-sided biopsy may be insufficient to rule out the diagnosis in as many as 40% of cases [59]. In practice, a trichrome stain, collagen type IV, and tenascin immunohistochemistry may aid in the detection of minimal subepithelial collagen thickening and entrapped capillaries [92, 93].

Collagenous colitis (H&E). The features include denuded surface epithelium and thickened subepithelial collagen table beneath the intercryptal epithelial surface. The inferior border of the collagen table entraps small capillaries (arrow) and inflammatory cells. The lamina propria is expanded by a lymphoplasmacytic infiltrate. The epithelium shows mucin depletion and reactive epithelial changes with increased intraepithelial lymphocytes (arrowhead)
Amyloidosis may on occasion be virtually indistinguishable from CC and performing a Congo red stain is helpful [94]. Progressive systemic sclerosis may be associated with a thickened fibrous band along basement membranes mimicking CC; however, the thickening extends along all basement membranes including crypts and additionally, fibrosis of the muscularis mucosae may be seen as well as intimal thickening of submucosal arteries [95].
Lymphocytic colitis
LC is characterized by an increase in surface and crypt intraepithelial lymphocytes (IELs). A threshold of greater than 20 per 100 enterocytes has been suggested (normal less than 5 per 100 enterocytes) (Fig. 8) [59]. The presence of epithelial injury including loss of the normal columnar shape, mucin depletion, and an attenuated or syncytial appearance of the surface epithelium is commonly noted [88]. However, regenerative changes with nuclear enlargement and a slight increase in mitotic activity are variable. The lamina propria has increased cellularity due to lymphocytes and plasma cells [92]. The architecture is preserved and there is no thickening of the subepithelial collagen table [88]. Enterocolic lymphocytic phlebitis, a rare process limited to the gut and characterized by lymphocytic inflammation of veins and venules, has been described in association with LC and is likely an iatrogenic manifestation of drugs [96].

Lymphocytic colitis (H&E). Increased IELs (arrowheads), mucin depletion, and reactive epithelial changes are seen. There is expansion of the lamina propria by a lymphoplasmacytic infiltrate
Phenotyping shows that IELs show a normal CD3+ CD8+ T cells expressing TCR-αβ in contrast to CD4+ T cells in the lamina propria [97]. The use of CD3 is of limited value if not to reveal several rarely overlooked cases [98].
Occasionally, confusing IBD-like histologic features may be seen in either LC or CC [78, 99, 100]. Cryptitis and crypt abscesses when present in MC (30% of CC and 38% of LC) are focal, involving less than 3 crypts per biopsy specimen in all affected cases [100], in contrast to active IBD where they may dominate the histologic picture. Crypt architectural changes and atrophy are uncommon (less than 10% of CC and LC) and when present are typically focal being limited to one portion of a single fragment [100]. Paneth cell metaplasia may be seen, being somewhat more common in CC where this may be associated with a more severe disease or prolonged course [100, 101].
Histologic variants of LC and CC
Several variants have been identified that appear to have similar clinical features and outcome as the more classic form (Table 1) [99].
“Paucicellular lymphocytic colitis” is characterized by a mild increase in lamina propria lymphoplasmacytic inflammation and with an increase in IELs (mean IELs of 11.1/100 vs. 29.3/100 for classic LC) [102]. Whether it is part of the morphologic spectrum of LC or not has been debated with several studies showing variance from typical LC not only in terms of prevalence but also immunopathogenesis with lack of CD25+FOXP3+ cells in the lamina propria when compared with classic LC [102, 103].
This variant is particularly difficult to differentiate from colonic epithelial lymphocytosis (Table 2) [104, 105]. A mild increase in IELs without obvious epithelial injury and increased lamina propria cellularity is nonspecific and may occur with medications, celiac disease (although LC may also occur with celiac disease), resolving infectious colitis, immunologic conditions (e.g., Hashimoto’s thyroiditis, common variable immune deficiency (CVID), autoimmune enteropathy, allergic conditions, immune deficiency), and IBD [104–106]. Brainerd diarrhea may also show intraepithelial lymphocytosis and is discussed elsewhere [107]. Resolving infectious colitis may show increased IELs and mild increase in lamina propria cellularity [3]. In general LC often shows a more diffuse and denser involvement with epithelial damage and different clinical presentation [104]. Knowledge of the clinical presentation, endoscopy, and culture findings will allow differentiation.
Pseudomembranes may be seen in CC (pseudomembranous CC), however, superimposed infections, especially C. difficile, and ischemic etiologies should be excluded.
Eosinophilic conditions affecting the colon
Eosinophilic disorders of the gut are classified as either primary or secondary (Table 3). Primary eosinophilic gastrointestinal disorders (EGID) are conditions that selectively affect the gastrointestinal tract in the absence of an identifiable cause for eosinophilia [108, 109]. These encompass eosinophilic esophagitis, eosinophilic gastritis, eosinophilic enteritis, eosinophilic gastroenteritis, and eosinophilic colitis [108, 109]. In the last two conditions, the colon can be involved together with other parts of the gut or exclusively.
Eosinophilic gastroenteritis
Eosinophilic gastroenteritis (EGE) is rare and characterized by selective eosinophilic infiltration of the stomach, small bowel, or both, and variable involvement of the esophagus and large bowel [109]. Eosinophilia may involve any layer(s) of the gastrointestinal tract and this forms the basis of subclassification into mucosal, muscularis, and serosal forms [109]. Any age group may be affected with most cases affecting adults in their thirties to fifties. There is no identifiable cause for eosinophilia. A subset of patients has an associated allergic component and may exhibit increased total IgE and food-specific IgE levels [109].
Mucosal and submucosal infiltration, which represents the most common form, is characterized by diarrhea, vomiting, weight loss, abdominal pain, malabsorption, iron deficiency anemia, and protein-losing enteropathy [108, 109]. Infiltration of the muscularis propria may lead to thickening of the bowel wall and obstruction [109]. Serosal involvement leads to exudative ascites and is often associated with higher peripheral eosinophilic counts [109]. The frequency of the various forms is reported as 57.5% for mucosal, 30% for muscular, and 12.5% for serosal disease [110]. Peripheral eosinophilia is variable and 25% of patients have normal eosinophil counts [110]. Atopy and allergies are associated in 25–75% of cases [108]. The evolution is variable with permanent resolution in some patients; however, recurrent relapses are the norm [108, 109].
The endoscopic features are nonspecific and include thickened folds, erythema, friability, nodularity, and abnormal peristalsis [108]. Involvement may be patchy and segmental. Endoscopy may be normal in muscular or serosal disease and imaging may be the only method to detect stenosis or thickening of the wall [108].
The histologic features are not specific and the diagnosis of EGE is a diagnosis of exclusion. Mucosal disease will show a dense eosinophilic infiltrate with a preserved architecture. However, there is no established threshold above which a diagnosis should be rendered [108]. Suggestive features include clustering or sheets of eosinophils, epithelial injury (e.g., mucin depletion), eosinophilic infiltration of epithelium, crypts, or glands, and muscularis mucosa or submucosa. Mast cells may be increased not surprisingly [109]. Mucosal biopsies may be normal in muscularis or serosal forms of disease.
Clinicopathological correlation and exclusion of other secondary causes of gastrointestinal eosinophilia are important (Table 3).
Eosinophilic colitis
Eosinophilic colitis is an uncommon disorder that selectively affects the colon in the absence of known cause of eosinophilia [109]. It more commonly affects infants and adolescents and most infantile cases are thought to be due to allergy to soy protein or cow’s milk protein [108, 111]. Yet interestingly, eosinophilic colitis has been reported to occur more often in infants that are exclusively breast fed [108, 109]. In contrast to EGE, eosinophilic colitis is usually a non-IgE-associated disease with some studies suggesting a T cell lymphocyte-mediated process but the exact underlying immunologic mechanism is not known [109].
Diarrhea, which can be bloody, is the classic presentation [108, 109]. Symptoms may include abdominal pain, anorexia, and weight loss. Most affected infants lack constitutional symptoms and are otherwise healthy. Peripheral blood eosinophilia and stool eosinophils may be present [108].
The colonoscopic features are nonspecific including erythema and loss of vascularity, friability, and nodularity [109].
Histologically, increased numbers of clustering eosinophils are noted within the lamina propria while the architecture is preserved (Fig. 9). Despite the absence of consensus criteria, a threshold of greater than 20 eosinophils per high power field (HPF) has been used [112]. Other useful features include infiltration of surface and crypt epithelium by eosinophils (Fig. 9) and infiltration of the muscularis mucosae by eosinophils.

Eosinophilic colitis (H&E). The lamina propria is expanded by eosinophil-rich infiltrate with clustering and degranulation of eosinophils. Eosinophils are also seen infiltrating the surface and crypt epithelium (arrows)
The features are not specific and it is a diagnosis of exclusion. Involvement of other segments of the gastrointestinal mucosa should suggest a diagnosis of EGE. Clinicopathological correlation and exclusion of other secondary causes of gastrointestinal eosinophilia are important (Table 3).
Of note, the so-called pericrypt eosinophilic enterocolitis is an unusual condition in which many patients suffer from systemic connective tissue disease and chronic watery diarrhea. The colonoscopy is normal and histologically, the eosinophils surround the deep crypts separating them from the muscularis mucosae and infiltrate muscularis mucosae [113].
Mastocytic enterocolitis
Mastocytic enterocolitis is a controversial entity characterized by chronic diarrhea with intermittent abdominal pain and increased mucosal mast cells (more than 20/HPF) [114]. At present, it is unclear if this represents a true entity or simply a morphologic reaction pattern [115]. No predisposing factor has been identified and it is not associated with systemic mastocytosis or cutaneous mastocytosis and serum tryptase is normal [114].
The age range is broad and a female predominance is reported [114]. The exact relationship of mastocytic colitis to diarrhea-predominant IBS is presently unknown. Colonoscopy and endoscopy are normal or show mild edema. A majority of patients respond to antihistamines or mast cell stabilizers or inhibitors of mediator release [116].
Histologically, the biopsies are essentially normal or show nonspecific mild and focal increase in mixed inflammatory cells or eosinophils in the lamina propria [114, 116]. Mast cell tryptase immunohistochemical stain demonstrates increased mast cells, with greater than 20 mast cells per HPF diagnostic of mastocytic enterocolitis, in the appropriate clinical setting [114, 116]. The normal number of mucosal mast cells is on average 13 per HPF [114, 116]. Although debated, it may be useful to perform a tryptase immunostain in patients with unexplained chronic diarrhea presenting like diarrhea-predominant IBS as this may identify a subset that could benefit from specific treatment [116].
Increased colonic mucosal mast cells may also be seen in ulcerative colitis [117], CC [118], gluten-sensitive enteropathy [119], radiation colitis [120], eosinophilic colitis [109], and all forms of irritable bowel syndrome [116]. Mast cell diseases that involve the gut include systemic mastocytosis and cutaneous mastocytosis with gastrointestinal symptoms. The mast cells are usually numerous forming confluent sheets and aggregates, associated with increased eosinophils, with distortion of the mucosa [121]. The presence of greater than 15 mast cells per HPF in a patient with systemic mastocytosis has been established as the threshold for a diagnosis of extracutaneous involvement [122].
Ischemic colitis
Ischemic colitis is a result of a hypoxic tissue injury with secondary necrosis and inflammation [123]. It is more common in the elderly (over 65 years) and women but younger patients may also be affected [124]. The true incidence is not known since many cases are probably never diagnosed or are misdiagnosed [124]. Ischemic colitis may result from various etiologies but can be broadly classified as non-occlusive and occlusive (Table 4). Non-occlusive conditions are the more common (e.g., cardiac shock or low cardiac output) and are often associated with preexisting conditions (e.g., atherosclerosis) [125]. The injury may be transmural (full thickness of the bowel wall), mural (injury involving the mucosa and to a variable extent the deeper layers of the bowel wall but falling short of full-thickness infarction), and mucosal (injury limited to the mucosa) [123, 125]. Reperfusion injury has been shown to result in a greater degree of tissue damage than that due to the actual ischemic period and is thought to be mediated by the formation of oxygen free radicals [126].
Any region of the colon can be involved including the rectum [125]; however, classically ‘watershed’ areas of the colon were thought to be most prone to ischemia [123, 125]. These are found in the region of the splenic flexure anastomosis between the vascular supply of the superior mesenteric and inferior mesenteric arteries, and the rectosigmoid anastomosis between the vascular supplies of the inferior mesenteric artery and the internal iliac artery. The rectum is thought to be relatively resistant to ischemia due to the dual splanchnic and systemic vascular supply [125]. The right colon is also prone to ischemia due to the smaller and less well-developed vasa recta compared to the left colon [127]. More recent evidence demonstrates that so-called watershed areas may not be the most common sites of involvement, with less than 14% of cases involving the splenic flexure alone and less than 5% involving the rectosigmoid [128]. Rather, the left colon is the most commonly involved region, followed by the transverse colon and splenic flexure, splenic flexure alone, right colon, and rectum in decreasing order of frequency [125]. This has led some to suggest that perhaps the traditional ‘watershed’ areas may in fact be ischemic resistant due to their ‘dual’ blood supply [128].
Clinically, acute onset mild, crampy abdominal pain, often with an urge to defecate, is the presenting symptom and passage of bright red or maroon-colored stool [125]. Peritonism is associated with transmural infarction [125]. Ischemic colitis may be classified clinically into gangrenous and non-gangrenous forms [125]. The latter is further subdivided into transient and reversible ischemic colitis (mucosa and submucosa involvement) and chronic, non-reversible ischemic colitis, which includes chronic and structuring disease [129]. Most patients have transient and reversible non-gangrenous ischemic colitis and only a minority (~10%) may subsequently present with strictures [125].
Recent evidence suggests that isolated right-sided ischemic colitis (IRIC) may represent a distinctive entity characterized by isolated involvement of the right colon and abdominal pain rather than hematochezia [130–132]. It is often associated with chronic renal failure (hemodialysis), coronary artery disease, and atrial fibrillation [130, 131]. Importantly, it is associated with a higher (fivefold) need for surgery and mortality (twofold) [130–132]. The reason for this worse outcome has been explained by the common blood supply of the right colon and small intestine via the superior mesenteric artery and frequent association of mesenteric ischemia with its greater need for surgery and higher mortality [130, 131].
Colonoscopic findings are variable. In early stages, the mucosa is pale and edematous with interspersed areas of hyperemia or petechial hemorrhage [133]. Later, segmental erythema, with or without ulceration and bleeding, may be seen [133]. With more advanced ischemia, submucosal edema and hemorrhage develop in the first 2–3 days and resolve quickly thereafter [125, 133]. In severe cases, the mucosa appears cyanotic, dusky, gray, or black [133]. Pseudopolyps and pseudomembranes may be noted [133]. Strictures, decreased haustrations, and mucosal granularity are features of chronic ischemia [134].
The microscopic features depend on the stage: (1) acute phase, with hemorrhage and necrosis; (2) reparative phase, with the formation of granulation tissue and fibrosis; (3) stricturing phase (and other complications) [135]. In early ischemia, the mucosal injury is predominantly superficial with varying degrees of flattening and degeneration of the upper crypt and surface epithelium, which may be sloughed. Intact crypt cells are flattened and show mucin depletion with hyperchromatic nuclei. Depending on the severity, the basal portions of crypts are viable (Fig. 10). There are also ectatic capillaries in which fibrin thrombi are seen. Edema, hemorrhage, and fibrin deposition may be seen and the lamina propria often develops a deeply eosinophilic hyalinized quality (Fig. 10). Neutrophils are sparse at the onset but a variable increased inflammatory infiltrate may develop. In severe cases, the crypts appear dilated, filled with mucin and inflammatory debris, and lined by attenuated epithelium. Acute inflammatory cells, plasma cells, and lymphocytes are present and crypt abscesses may be seen. Complete epithelial loss leading to empty crypt outlines may be observed. Full-thickness mucosal necrosis with mucosal sloughing and ulceration is seen in severe and persisting ischemia. The submucosa is markedly edematous with vascular congestion and mixed inflammation. Vascular thrombi and endophlebitis may be seen beneath the ulcerated mucosa but these should not be construed as the cause of ischemia unless these are distant in the non-ulcerated mucosa.

Ischemic colitis (H&E). Necrosis of superficial crypts with viable crypt bases is present (arrows). The changes are pauci-inflammatory and characteristic hyalinization of the lamina propria with congestion and red cell extravasation (arrowheads) is seen. There is edema of the submucosa
A reparative phase usually follows the acute phase but they may be seen concurrently. In mild cases, the mucosa returns to normal with no significant architectural distortion. In severe cases, mucosal fibrosis occurs and re-epithelialization occurs from residual viable cells at the base of surviving crypts or from ingrowth of surface epithelium but a normal mucosal pattern may not be restored with distorted glands embedded within fibrous lamina propria containing hemosiderin-laden histiocytes. Large ulcers may heal leaving an atrophic area of re-epithelialized flat surface without crypts. Pyloric gland and Paneth cell metaplasia, endocrine cell hyperplasia, or colitis cystica profunda may develop. Fibrosis may ultimately lead to strictures.
The differential diagnosis of ischemic colitis includes pseudomembranous colitis since pseudomembranes can develop when mucosal injury is limited to the superficial half of the mucosa [24]. Ischemic colitis should also be distinguished from infectious colitis with “withering” crypts and degeneration of the upper crypt epithelium. It should be remembered that certain infectious colitides have an ischemic component as part of their pathogenesis (e.g., EHEC), thus resulting in overlapping features [12, 14].
In chronic ischemia, distortion of the mucosal architectural may raise the possibility of IBD. Chronic radiation colitis may also resemble chronic ischemia.
Diverticular disease-associated colitis
Diverticular disease is a condition occurring in at least 50% of the population over the age of 60 in the West [136]. It is associated with a low-fiber diet, increased colonic transit time, chronic constipation, and straining with increased intraluminal pressure [136]. It is characterized by outpouchings of the colonic mucosa with or without residual wisps of muscularis mucosae but lacking a formal muscular layer. The left colon is more commonly affected in Western patients [136]. The complications of diverticular disease include hemorrhage, inflammation with abscess or fistulae, stricture with mechanical large bowel obstruction, and colonic perforation leading to peritonitis [136, 137]. In addition, tonic muscular contraction gives rise to redundancy of surface mucosal folds prone to prolapse-type/ischemic injury [136, 137].
In less than 2% of the cases, diverticular disease-associated chronic colitis can develop in the segment of colon affected [137]. Its pathogenesis has not been elucidated [136].
Colonoscopic examination may show erythematous and granular mucosa similar to IBD. In severe cases, linear ulceration and a cobblestone mucosal pattern resembling Crohn’s disease may be seen [136, 138].
Histologically, the mucosa shows changes that are indistinguishable from IBD including architectural distortion, Paneth cell metaplasia, increase in lamina propria mononuclear infiltration, basal lymphoid aggregates and plasmacytosis, cryptitis, crypt abscesses, surface epithelial sloughing, and granulomatous cryptitis [136, 138, 139]. Non-necrotizing granulomas may be seen involving the mucosa and submucosa of diverticula, but are not concentrated around them [138]. Non-necrotizing granulomas may also be seen in pericolonic lymph nodes [138]. Transmural lymphoid aggregates similar to Crohn’s disease may be seen, most often in the pericolonic adipose tissue [138]. Occlusive arterial fibroplasia, lymphohistiocytic and granulomatous vasculitis are rarely reported [138].
Clues allowing the differentiation from ulcerative colitis include the restriction of inflammatory changes to the segment involved by diverticular disease with a normal rectum and colon proximal to the involved segment [136, 140]. However, ulcerative colitis with rectal sparing may still enter the differential diagnosis [139, 141]. In cases demonstrating Crohn’s disease-like features, a diagnosis of Crohn’s disease should be avoided unless there is evidence of disease elsewhere either concurrently or on follow-up [136, 138].
Mucosal prolapse (MP)/solitary rectal ulcer syndrome (SRUS)
SRUS is a rare condition associated with disorder of defecation typically affecting young adults. The typical presentation includes rectal bleeding and passage of mucus, constipation, tenesmus, straining, or diarrhea [142, 143]. Several colonoscopic features may be seen including typical solitary ulcer (35%) or multiple ulcers (22%), polypoid lesions (25%), or hyperemic mucosa (18%) [144]. The lesions are most often found 5–10 cm from the anal margin on the anterior or anterolateral wall of the rectum and circumferential changes can be seen [145].
The pathogenesis of SRUS is believed to involve rectal MP with paradoxical contraction of the pelvic floor opposing the downward forces of defecation [146, 147].
The histologic features include thickening of the mucosa with elongation, increased tortuosity, dilatation and distortion of the crypts, particularly at the base (Fig. 11) [148, 149]. The crypt bases may become pointed and on cross section (‘diamond-shaped’) [150]. There is commonly surface erosion with fibrin exudate and neutrophils [148], resembling the type I lesion of pseudomembranous colitis. Evidence of ischemia with degeneration of crypts may be present [148, 149]. The surface epithelium can show regenerative changes as well as hypertrophied goblet cells [148, 151]. The lamina propria is edematous with congested superficial capillaries and a variable proliferation of fibroblasts [148]. Pauci-inflammatory changes are seen. The muscularis mucosae is hypertrophied with splaying of variably orientated fibers extending upward between crypts. Localized colitis cystica profunda can be seen [148].

Mucosal prolapse (H&E). The lesion is characterized by thickening of the mucosa, elongation and increased tortuosity, dilatation and distortion of the crypts, and characteristic splaying of the muscularis mucosae
Similar histologic changes can occur associated with other conditions that predispose to MP, i.e., protruding lesions/masses, redundant mucosal folds in the contracted colon of diverticular disease (so-called crescentic fold colitis) and adjacent to stomal or diverticular openings [149, 152, 153].
The differential diagnosis includes ischemia, early pseudomembranous colitis (type I lesion), and IBD [148, 154]. Other polypoid lesions with MP features include inflammatory cloacogenic polyp, inflammatory cap polyp, and inflammatory myoglandular polyp [154]. SRUS/MP may on occasion mimic carcinoma endoscopically [145].
Colitis cystica profunda (CCP)
CCP describes a distinct histologic pattern characterized by the displacement of glands below the muscularis mucosae, often accompanied by lamina propria. The features may be seen in MP syndromes, IBD, and radiation injury [149, 155, 156].
CCP is thought to be due to misplaced glands derived from either downgrowth of epithelium during regeneration in the healing of deeply penetrating ulceration or chronic colonic ischemia [155]. Misplaced glands are well-circumscribed lacking an infiltrative growth pattern and neoplastic cytological features, often with a rim of surrounding lamina propria.
Three patterns of involvement are recognized: (1) a mass lesion, usually in the rectum, which may mimic an obstructing carcinoma typically due to SRUS; (2) segmental involvement following healing of segment Crohn’s disease or radiation damage; (3) diffuse involvement of the bowel, e.g., following healing ulceration in bacillary dysentery or ulcerative colitis [157, 158].
Histologically, CCP shows mucin-filled cysts lined by benign colonic epithelium beneath the muscularis mucosae with or without rupture and extravasation of mucin. Some cysts may have an incomplete lining. There may be variable features of mucosal injury. The displaced epithelium is usually, although not invariably, associated with lamina propria. The epithelium does not show dysplastic features although it may be reactive or regenerative. Histologic features related to the underlying condition including radiation damage, hemorrhage, and hemosiderin deposition can be seen.
The most important differential diagnosis is distinguishing the findings from invasive adenocarcinoma including mucinous adenocarcinoma [158]. The presence of dysplastic complex or papillary glandular structures, irregular or angulated glandular profiles with a haphazard infiltrative pattern, and stromal desmoplasia are features favoring adenocarcinoma.
Pneumatosis coli
Pneumatosis coli is characterized by collection of gas in the wall of the colon [159]. The pathogenesis of pneumatosis coli is not known; however, analysis of the cystic gas suggests three potential sources, namely intraluminal, pulmonary, or bacterial [160, 161]. More common in adults, it has been described in association with a wide variety of conditions including traumatic, mechanical, inflammatory, autoimmune, collagen vascular disease, infectious and drug-induced etiologies that share in common compromised mucosal integrity and/or increased intraluminal pressure increasing the likelihood of gas penetrating the intestinal wall [159]. An association with pulmonary disease, especially chronic obstructive pulmonary disease, is also seen in many cases [162].
Pneumatosis coli may be asymptomatic or accompanied by constipation, diarrhea, abdominal pain, obstruction, intussusception, or volvulus [158, 163, 164]. Symptoms and features referable to the underlying condition may be noted [159]. The presence of gas within the bowel wall may be demonstrated on imaging [165]. On colonoscopy, soft rounded cystic-nodular lesions, which may mimic polyps or polyposis, can be seen [159, 166].
Histologically, the diagnosis is obvious on resections with the presence of air cysts often within the submucosa lined by histiocytes and giant cells (Fig. 12); however, cysts may be present within all layers of the bowel wall [159]. On mucosal biopsies, the diagnosis may be more challenging. Architectural distortion, cryptitis, crypt abscesses, chronic inflammation, and granulomas may be detected mimicking IBD, particularly Crohn’s disease [159, 161, 167]. Crypt dilation and partial crypt rupture, with formation of intramucosal and submucosal cysts associated with histiocytic and giant cell aggregates, may be seen [161]. However, these are often disrupted or collapsed and the arrangement of giant cells along a cleft-like irregular branching space is critical to distinguishing it from granulomatous inflammation, which it can mimic [159]. Other useful clues include the presence of pseudolipomatosis or larger submucosal air spaces [159]. The latter may resemble adipocytes; however, these are too large or confluent to be adipocytes and occasionally may surround normal structures such as nerves [159].

Pneumatosis coli (H&E). There are submucosal air-filled spaces with multinucleated giant cells at the periphery (arrowheads)
Diversion colitis
Diversion colitis is an iatrogenic inflammatory disorder that develops consequent to defunctioning segments of large bowel with diversion of the fecal stream, most commonly due to ileostomy or colostomy [168]. The abnormalities can become apparent within 3 months of diversion [169]. It is widely held that these changes are due to alteration in the luminal flora with a resulting lack of short chain fatty acids trophic to the colonic mucosa [168, 170]. Alternatively, an underlying vascular or vasogenic abnormality has been proposed [171].
The patients with diversion colitis may be asymptomatic or present with mucoid or bloody discharge and crampy abdominal pain [168]. On colonoscopy, there may be erythema, petechial hemorrhage, mucosal friability, mucosal nodularity, sometimes surrounded by aphthous ulcers.
Two characteristic histologic findings can be observed, one is the development of marked lymphoid hyperplasia with or without focal overlying erosions, and various degrees of architectural changes with increase in the lamina propria chronic inflammatory cells. The acute inflammation with focal neutrophil infiltration in the lamina propria, cryptitis, and crypt abscesses is less commonly seen. Inflammatory pseudopolyps may develop. The inflammation in diversion proctocolitis may mimic IBD [172], but the distinction is made by the absence of significant crypt distortion [173] and the lymphoid hyperplasia is characteristic. Exacerbation of inflammation with unusual features may be seen in diverted large bowel of patients with preexisting diagnosis of ulcerative colitis including fissuring ulceration and granulomas that should not be construed as evidence of Crohn’s disease [174]. Microcarcinoids have also been reported in the diverted rectum in ulcerative colitis [175]. In contrast, the histological appearances of diverted Crohn’s disease typically show ‘burnt-out disease’ with fibrosis and a lack of active inflammation [168, 174]. Granulomas become effete with hyalinization [176].
Radiation colitis
Acute radiation toxicity and long-term consequences of irradiation can be observed. Radiation injury to the gut may occur following total doses below 40 Gy; however, the incidence of significant injury increases when this exceeds 50 Gy [177]. Acute radiation colitis is usually seen within 2 weeks following irradiation. The symptoms of acute radiation include diarrhea, mucoid discharge, bleeding, tenesmus, urgency, and incontinence [178, 179]. On colonoscopy, the mucosa appears edematous with loss of vascular pattern. Granularity, hemorrhage (Fig. 13a), and multiple ulcers may be seen [178, 180]. Radiation-induced chronic colitis usually develops 6 months to 5 years following irradiation but may be delayed for decades [178, 181].

Radiation colitis. Colonoscopy showing mucosal congestion with hemorrhage (a). Chronic radiation colitis characterized by distortion and dilation of crypts together with atrophy and crypt loss with reduced crypt density and increased intercryptal distance (b H&E). The surface epithelium is mucin depleted and cuboidal (arrowhead). Prominent lamina propria fibrosis is present with telangiectasia (arrows). The muscularis muscosae is thickened. Atypical ‘radiation’ fibroblasts are appreciated at higher magnification characterized by hyperchromatic smudged nuclei (arrow) (c H&E). Submucosal fibrosis and vascular hyalinization (arrow) is seen and mucosal telangiectasia is once again noted (arrowhead) (d H&E)
On mucosal biopsy, acute radiation changes include crypt cell injury with nuclear atypia, mucin depletion, and loss of polarity, crypt abscesses, crypt loss, acute ulceration, and mucosal sloughing [179]. Apoptotic bodies and GVHD-like ‘exploding crypts’ may be seen [178, 179]. The lamina propria is edematous and acute inflammatory cells are present, often with a prominent eosinophilic infiltrate and eosinophilic crypt abscesses can be observed [179]. Telangiectasia is observed and vascular fibrinoid necrosis and mild vascular fibrosis may be seen [178, 179, 182]. Submucosal edema often with a myxoid quality is common [183]. Histologic features of ischemia may be observed. Resolution occurs within 1 month but some changes may persist up to 3 months [184].
Delayed changes include hyalinized vessels and stromal fibrosis along with vascular ectasia and lymphangiectasia (Fig. 13b, c) [178, 179]. Marked hyalinization of submucosal vessels (Fig. 13d) may be seen along with neuronal proliferation [182]. In some cases, Congo red may be needed to exclude amyloidosis. Arterioles often show intimal proliferation, sometimes with foamy endothelial cells [182]. Chronic architectural distortion with variable atrophy, loss of goblet cells, and Paneth cell metaplasia may be seen [179, 185]. Epithelial atypia may be noted characterized by nuclear enlargement and smudged hyperchromatic nuclei with preserved nuclear to cytoplasmic ratio. Thickening of the collagen layer beneath the surface and crypt epithelium may be seen that should not be mistaken for CC. The muscularis mucosae is thickened and splayed and cystica profunda may be seen [155]. Mucosal erosion and ulcers may occur. Atypical “radiation” fibroblasts with enlarged hyperchromatic nuclei and basophilic cytoplasm are present in the lamina propria [178].
Focal active colitis (FAC)
FAC is a nonspecific inflammatory lesion characterized by focal cryptitis or crypt abscesses in the absence of any other significant microscopic abnormality [186]. It is not an uncommon diagnosis, estimated to be diagnosed approximately 50 times per year in the average general hospital practice [187], and may be more common in females [186, 187]. Patients may be asymptomatic; however, most commonly they have diarrhea with or without abdominal pain [186–189]. FAC has been found to be associated with multiple conditions (Table 5) [186–190]. The colonoscopic appearances include patchy erythema, or nonspecific findings including possible aphthous ulcers or vascular ectasia, subepithelial hemorrhage but may be essentially normal [186]. FAC may also be an incidental biopsy finding [186–189].
Early studies found infections/ASLC to be most commonly associated with FAC in adults and pediatric cases (Table 5). Although early data found no cases of IBD in adults with a median follow-up of 22 months [186], subsequent studies have shown up to 13% of adults subsequently develop Crohn’s disease with a higher proportion in pediatric cases (27.6%) [188, 189]. More recent data found 16% of adults develop IBD between 18 months to 6 years following a diagnosis of FAC, mostly Crohn’s disease, but may also develop ulcerative colitis or IBD unclassified [187]. Drugs, particularly NSAIDS, are an important cause of FAC and this needs to be considered in the etiologic diagnosis [186, 191]. A single study using PCR demonstrated in a series of 110 patients that 19% could be etiologically traced to Campylobacter jejuni [192]. In a proportion of patients, the pattern is associated with antibiotic-related colitis or ischemic colitis [186, 189]. FAC may also be seen with sodium phosphate bowel preparations [193, 194].
Microscopically, FAC is characterized by patchy isolated evidence of focal crypt injury ranging from a single crypt abscess or focus of cryptitis to multiple discrete foci of cryptitis or crypt abscesses (less than 50% of the biopsy specimen) (Fig. 14) [186, 188, 189]. There is characteristically no architectural alteration. Apoptosis may be common as well. The pattern of ‘basal FAC’, especially if associated with apoptosis, may be predictive of a drug etiology [187].

Focal active colitis (H&E). Focal isolated cryptitis and crypt abscess formation (arrow) in an otherwise unremarkable colonic mucosa
The diagnosis of FAC should lead to the exclusion of the various etiologies (Table 5). In practical terms, FAC should also be distinguished from focal chronic active colitis, in which architectural and other features of chronicity are observed.
Conclusion
Even after a diagnosis of IBD is eliminated, the diagnosis of inflammatory conditions of the colon remains a significant challenge. Successful management of patients necessitates a close working relationship between the endoscopist and the pathologist who both need to consider a wide differential diagnosis.
References
Kumar NB, Nostrant TT, Appelman HD. The histopathologic spectrum of acute self-limited colitis (acute infectious-type colitis). Am J Surg Pathol. 1982;6:523–9.
Navaneethan U, Giannella RA. Mechanisms of infectious diarrhea. Nat Clin Pract Gastroenterol Hepatol. 2008;5:637–47.
Lamps LW. Infective disorders of the gastrointestinal tract. Histopathology. 2007;50:55–63.
Navaneethan U, Giannella RA. Infectious colitis. Curr Opin Gastroenterol. 2011;27:66–71.
Prescott RJ, Harris M, Banerjee SS. Fungal infections of the small and large intestine. J Clin Pathol. 1992;45:806–11.
Schumacher G, Sandstedt B, Mollby R, Kollberg B. Clinical and histologic features differentiating non-relapsing colitis from first attacks of inflammatory bowel disease. Scand J Gastroenterol. 1991;26:151–61.
Mantzaris GJ. Endoscopic diagnosis of infectious colitis. Ann Gastroenterol. 2007;20:71–4.
Surawicz CM, Haggitt RC, Husseman M, McFarland LV. Mucosal biopsy diagnosis of colitis: acute self-limited colitis and idiopathic inflammatory bowel disease. Gastroenterology. 1994;107:755–63.
Carpenter HA, Talley NJ. The importance of clinicopathological correlation in the diagnosis of inflammatory conditions of the colon: histological patterns with clinical implications. Am J Gastroenterol. 2000;95:878–96.
Sherman PM, Ossa JC, Wine E. Bacterial infections: new and emerging enteric pathogens. Curr Opin Gastroenterol. 2010;26:1–4.
Tarr PI. Escherichia coli O157:H7: clinical, diagnostic, and epidemiological aspects of human infection. Clin Infect Dis. 1995;20:1–8 (quiz 9–10).
Griffin PM, Olmstead LC, Petras RE. Escherichia coli O157:H7-associated colitis. A clinical and histological study of 11 cases. Gastroenterology. 1990;99:142–9.
Bielaszewska M, Karch H. Consequences of enterohaemorrhagic Escherichia coli infection for the vascular endothelium. Thromb Haemost. 2005;94:312–8.
Kelly J, Oryshak A, Wenetsek M, Grabiec J, Handy S. The colonic pathology of Escherichia coli O157:H7 infection. Am J Surg Pathol. 1990;14:87–92.
Ananthakrishnan AN. Clostridium difficile infection: epidemiology, risk factors and management. Nat Rev Gastroenterol Hepatol. 2011;8:17–26.
Freeman J, Bauer MP, Baines SD, Corver J, Fawley WN, Goorhuis B, et al. The changing epidemiology of Clostridium difficile infections. Clin Microbiol Rev. 2010;23:529–49.
Drudy D, Harnedy N, Fanning S, O’Mahony R, Kyne L. Isolation and characterisation of toxin A-negative, toxin B-positive Clostridium difficile in Dublin, Ireland. Clin Microbiol Infect. 2007;13:298–304.
Hurley BW, Nguyen CC. The spectrum of pseudomembranous enterocolitis and antibiotic-associated diarrhea. Arch Intern Med. 2002;162:2177–84.
Price AB, Davies DR. Pseudomembranous colitis. J Clin Pathol. 1977;30:1–12.
Bartlett JG. Historical perspectives on studies of Clostridium difficile and C. difficile infection. Clin Infect Dis. 2008;46(Suppl 1):S4–11.
Price AB. Histopathology of clostridial gut disease in man. In: Borriello SP, editor. Clostridia in gastrointestinal disease. Boca Raton: CRC; 1985. p. 177.
Schiffman R. Signet-ring cells associated with pseudomembranous colitis. Am J Surg Pathol. 1996;20:599–602.
Wang K, Weinrach D, Lal A, Musunuri S, Ramirez J, Ozer O, et al. Signet-ring cell change versus signet-ring cell carcinoma: a comparative analysis. Am J Surg Pathol. 2003;27:1429–33.
Dignan CR, Greenson JK. Can ischemic colitis be differentiated from C. difficile colitis in biopsy specimens? Am J Surg Pathol. 1997;21:706–10.
Papaconstantinou HT, Thomas JS. Bacterial colitis. Clin Colon Rectal Surg. 2007;20:18–27.
El-Maraghi NR, Mair NS. The histopathology of enteric infection with Yersinia pseudotuberculosis. Am J Clin Pathol. 1979;71:631–9.
Ebringer A, Wilson C. HLA molecules, bacteria and autoimmunity. J Med Microbiol. 2000;49:305–11.
Matsumoto T, Iida M, Matsui T, Sakamoto K, Fuchigami T, Haraguchi Y, et al. Endoscopic findings in Yersinia enterocolitica enterocolitis. Gastrointest Endosc. 1990;36:583–7.
Gleason TH, Patterson SD. The pathology of Yersinia enterocolitica ileocolitis. Am J Surg Pathol. 1982;6:347–55.
Lamps LW, Havens JM, Gilbrech LJ, Dube PH, Scott MA. Molecular biogrouping of pathogenic Yersinia enterocolitica: development of a diagnostic PCR assay with histologic correlation. Am J Clin Pathol. 2006;125:658–64.
Misra SP, Misra V, Dwivedi M, Gupta SC. Colonic tuberculosis: clinical features, endoscopic appearance and management. J Gastroenterol Hepatol. 1999;14:723–9.
Tandon HD, Prakash A. Pathology of intestinal tuberculosis and its distinction from Crohn’s disease. Gut. 1972;13:260–9.
Pulimood AB, Amarapurkar DN, Ghoshal U, Phillip M, Pai CG, Reddy DN, et al. Differentiation of Crohn’s disease from intestinal tuberculosis in India in 2010. World J Gastroenterol. 2011;17:433–43.
Alvares JF, Devarbhavi H, Makhija P, Rao S, Kottoor R. Clinical, colonoscopic, and histological profile of colonic tuberculosis in a tertiary hospital. Endoscopy. 2005;37:351–6.
Gaffney EF, Condell D, Majmudar B, Nolan N, McDonald GS, Griffin M, et al. Modification of caecal lymphoid tissue and relationship to granuloma formation in sporadic ileocaecal tuberculosis. Histopathology. 1987;11:691–704.
van Mook WN, Koek GH, van der Ven AJ, Ceelen TL, Bos RP. Human intestinal spirochaetosis: any clinical significance? Eur J Gastroenterol Hepatol. 2004;16:83–7.
Korner M, Gebbers JO. Clinical significance of human intestinal spirochetosis—a morphologic approach. Infection. 2003;31:341–9.
Carr NJ, Mahajan H, Tan KL, Sharma R. The histological features of intestinal spirochetosis in a series of 113 patients. Int J Surg Pathol. 2010;18:144–8.
Lee FD, Kraszewski A, Gordon J, Howie JG, McSeveney D, Harland WA. Intestinal spirochaetosis. Gut. 1971;12:126–33.
Haque R, Huston CD, Hughes M, Houpt E, Petri WA Jr. Amebiasis. N Engl J Med. 2003;348:1565–73.
McMillan A, Gilmour HM, McNeillage G, Scott GR. Amoebiasis in homosexual men. Gut. 1984;25:356–60.
Herbinger KH, Fleischmann E, Weber C, Perona P, Loscher T, Bretzel G. Epidemiological, clinical, and diagnostic data on intestinal infections with Entamoeba histolytica and Entamoeba dispar among returning travelers. Infection. 2011;39:527–35.
Leung J, Chin A. Amebic colitis. Gastrointest Endosc. 2002;56:732.
Prathap K, Gilman R. The histopathology of acute intestinal amebiasis. A rectal biopsy study. Am J Pathol. 1970;60:229–46.
Mitchell LM, Mora LO. Amebiasis simulating idiopathic inflammatory disease of the colon. J Miss State Med Assoc. 1971;12:469–82.
Yamamoto-Furusho JK, Torijano-Carrera E. Intestinal protozoa infections among patients with ulcerative colitis: prevalence and impact on clinical disease course. Digestion. 2010;82:18–23.
Schuster FL, Ramirez-Avila L. Current world status of Balantidium coli. Clin Microbiol Rev. 2008;21:626–38.
Hulman G, Taylor LA. Entamoeba histolytica: demonstration by PAS/Martius yellow technique. Med Lab Sci. 1987;44:396–7.
Galiatsatos P, Shrier I, Lamoureux E, Szilagyi A. Meta-analysis of outcome of cytomegalovirus colitis in immunocompetent hosts. Dig Dis Sci. 2005;50:609–16.
Baroco AL, Oldfield EC. Gastrointestinal cytomegalovirus disease in the immunocompromised patient. Curr Gastroenterol Rep. 2008;10:409–16.
Le ST, Lee SS, Prideaux L, Block AA, Moore GT. Primary cytomegalovirus ileitis complicated by massive gastrointestinal haemorrhage in a patient with steroid refractory Crohn’s disease. Intern Med J. 2010;40:788–91.
Hunt NAC, Jewell D, Mortensen N, Warren B. Cytomegalovirus infection complicating ulcerative colitis. CPD Bull Cell Pathol. 1999;1:72.
Cheung AN, Ng IO. Cytomegalovirus infection of the gastrointestinal tract in non-AIDS patients. Am J Gastroenterol. 1993;88:1882–6.
Francis ND, Boylston AW, Roberts AH, Parkin JM, Pinching AJ. Cytomegalovirus infection in gastrointestinal tracts of patients infected with HIV-1 or AIDS. J Clin Pathol. 1989;42:1055–64.
Warren BF, Edwards CM, Travis SP. ‘Microscopic colitis’: classification and terminology. Histopathology. 2002;40:374–6.
Chande N, Driman DK, Reynolds RP. Collagenous colitis and lymphocytic colitis: patient characteristics and clinical presentation. Scand J Gastroenterol. 2005;40:343–7.
Giardiello FM, Lazenby AJ, Bayless TM, Levine EJ, Bias WB, Ladenson PW, et al. Lymphocytic (microscopic) colitis. Clinicopathologic study of 18 patients and comparison to collagenous colitis. Dig Dis Sci. 1989;34:1730–8.
Baert F, Wouters K, D’Haens G, Hoang P, Naegels S, D’Heygere F, et al. Lymphocytic colitis: a distinct clinical entity? A clinicopathological confrontation of lymphocytic and collagenous colitis. Gut. 1999;45:375–81.
Pardi DS, Kelly CP. Microscopic colitis. Gastroenterology. 2011;140:1155–65.
El-Matary W, Girgis S, Huynh H, Turner J, Diederichs B. Microscopic colitis in children. Dig Dis Sci. 2010;55:1996–2001.
Bogomoletz WV. Collagenous, microscopic and lymphocytic colitis. An evolving concept. Virchows Arch. 1994;424:573–9.
Bohr J, Larsson LG, Eriksson S, Jarnerot G, Tysk C. Colonic perforation in collagenous colitis: an unusual complication. Eur J Gastroenterol Hepatol. 2005;17:121–4.
Jarnerot G, Tysk C, Bohr J, Eriksson S. Collagenous colitis and fecal stream diversion. Gastroenterology. 1995;109:449–55.
Varghese L, Galandiuk S, Tremaine WJ, Burgart LJ. Lymphocytic colitis treated with proctocolectomy and ileal J-pouch-anal anastomosis: report of a case. Dis Colon Rectum. 2002;45:123–6.
Green PH, Yang J, Cheng J, Lee AR, Harper JW, Bhagat G. An association between microscopic colitis and celiac disease. Clin Gastroenterol Hepatol. 2009;7:1210–6.
Matteoni CA, Goldblum JR, Wang N, Brzezinski A, Achkar E, Soffer EE. Celiac disease is highly prevalent in lymphocytic colitis. J Clin Gastroenterol. 2001;32:225–7.
Freeman HJ. Collagenous colitis as the presenting feature of biopsy-defined celiac disease. J Clin Gastroenterol. 2004;38:664–8.
Marteau P, Lavergne-Slove A, Lemann M, Bouhnik Y, Bertheau P, Becheur H, et al. Primary ileal villous atrophy is often associated with microscopic colitis. Gut. 1997;41:561–4.
Sapp H, Ithamukkala S, Brien TP, Ayata G, Shaz B, Dorfman DM, et al. The terminal ileum is affected in patients with lymphocytic or collagenous colitis. Am J Surg Pathol. 2002;26:1484–92.
Lewis FW, Warren GH, Goff JS. Collagenous colitis with involvement of terminal ileum. Dig Dis Sci. 1991;36:1161–3.
O’Brien BH, McClymont K, Brown I. Collagenous ileitis: a study of 13 cases. Am J Surg Pathol. 2011;35:1151–7.
Leung ST, Chandan VS, Murray JA, Wu TT. Collagenous gastritis: histopathologic features and association with other gastrointestinal diseases. Am J Surg Pathol. 2009;33:788–98.
Koskela RM, Niemela SE, Lehtola JK, Bloigu RS, Karttunen TJ. Gastroduodenal mucosa in microscopic colitis. Scand J Gastroenterol. 2011;46:567–76.
Beaugerie L, Pardi DS. Review article: drug-induced microscopic colitis—proposal for a scoring system and review of the literature. Aliment Pharmacol Ther. 2005;22:277–84.
Beaugerie L, Luboinski J, Brousse N, Cosnes J, Chatelet FP, Gendre JP, et al. Drug induced lymphocytic colitis. Gut. 1994;35:426–8.
Fernandez-Banares F, Esteve M, Espinos JC, Rosinach M, Forne M, Salas A, et al. Drug consumption and the risk of microscopic colitis. Am J Gastroenterol. 2007;102:324–30.
Moneret-Vautrin DA, Delvaux M, Labouyrie E, El Gueddari Y, Morisset M, Hochard H. Collagenous colitis: possible link with isotretinoin. Eur Ann Allergy Clin Immunol. 2006;38:124–5.
Geboes K. Lymphocytic, collagenous and other microscopic colitides: pathology and the relationship with idiopathic inflammatory bowel diseases. Gastroenterol Clin Biol. 2008;32:689–94.
Jarnerot G, Hertervig E, Granno C, Thorhallsson E, Eriksson S, Tysk C, et al. Familial occurrence of microscopic colitis: a report on five families. Scand J Gastroenterol. 2001;36:959–62.
Fine KD, Do K, Schulte K, Ogunji F, Guerra R, Osowski L, et al. High prevalence of celiac sprue-like HLA-DQ genes and enteropathy in patients with the microscopic colitis syndrome. Am J Gastroenterol. 2000;95:1974–82.
Koskela RM, Karttunen TJ, Niemela SE, Lehtola JK, Ilonen J, Karttunen RA. Human leucocyte antigen and TNFalpha polymorphism association in microscopic colitis. Eur J Gastroenterol Hepatol. 2008;20:276–82.
Jensen L, Munck LK. Elementary diet reduces diarrhoea associated with microscopic colitis. Scand J Gastroenterol. 2005;40:1495–6.
Burgel N, Bojarski C, Mankertz J, Zeitz M, Fromm M, Schulzke JD. Mechanisms of diarrhea in collagenous colitis. Gastroenterology. 2002;123:433–43.
Lee E, Schiller LR, Vendrell D, Santa Ana CA, Fordtran JS. Subepithelial collagen table thickness in colon specimens from patients with microscopic colitis and collagenous colitis. Gastroenterology. 1992;103:1790–6.
Beaugerie L, Napoleon B, Ponchon T, Boyer J, Canard JM, Dalbies P, et al. Guidelines of the French Society for Digestive Endoscopy (SFED). Role of endoscopy in microscopic colitis. Endoscopy. 2005;37:97–8.
Koulaouzidis A, Saeed AA. Distinct colonoscopy findings of microscopic colitis: not so microscopic after all? World J Gastroenterol. 2011;17:4157–65.
Cimmino DG, Mella JM, Pereyra L, Luna PA, Casas G, Caldo I, et al. A colorectal mosaic pattern might be an endoscopic feature of collagenous colitis. J Crohns Colitis. 2010;4:139–43.
Robert ME. Microscopic colitis: pathologic considerations, changing dogma. J Clin Gastroenterol. 2004;38:S18–26.
Nyhlin N, Bohr J, Eriksson S, Tysk C. Systematic review: microscopic colitis. Aliment Pharmacol Ther. 2006;23:1525–34.
Otegbayo JA, Otegbeye FM, Rotimi O. Microscopic colitis syndrome—a review article. J Natl Med Assoc. 2005;97:678–82.
Williams JJ, Beck PL, Andrews CN, Hogan DB, Storr MA. Microscopic colitis—a common cause of diarrhoea in older adults. Age Ageing. 2010;39:162–8.
Tagkalidis P, Bhathal P, Gibson P. Microscopic colitis. J Gastroenterol Hepatol. 2002;17:236–48.
Muller S, Neureiter D, Stolte M, Verbeke C, Heuschmann P, Kirchner T, et al. Tenascin: a sensitive and specific diagnostic marker of minimal collagenous colitis. Virchows Arch. 2001;438:435–41.
Groisman GM, Lachter J, Vlodavsky E. Amyloid colitis mimicking collagenous colitis. Histopathology. 1997;31:201–2.
Halaby IA, Rantis PC, Vernava AM 3rd, Longo WE. Collagenous colitis: pathogenesis and management. Dis Colon Rectum. 1996;39:573–8.
Jain R, Chetty R. Enterocolic lymphocytic phlebitis and lymphocytic colitis: drug-related coexistent pathology. Int J Colorectal Dis. 2009;24:473–4.
Mosnier JF, Larvol L, Barge J, Dubois S, De La Bigne G, Henin D, et al. Lymphocytic and collagenous colitis: an immunohistochemical study. Am J Gastroenterol. 1996;91:709–13.
Mohamed N, Marais M, Bezuidenhout J. Microscopic colitis as a missed cause of chronic diarrhea. World J Gastroenterol. 2011;17:1996–2002.
Chang F, Deere H, Vu C. Atypical forms of microscopic colitis: morphological features and review of the literature. Adv Anat Pathol. 2005;12:203–11.
Ayata G, Ithamukkala S, Sapp H, Shaz BH, Brien TP, Wang HH, et al. Prevalence and significance of inflammatory bowel disease-like morphologic features in collagenous and lymphocytic colitis. Am J Surg Pathol. 2002;26:1414–23.
Goff JS, Barnett JL, Pelke T, Appelman HD. Collagenous colitis: histopathology and clinical course. Am J Gastroenterol. 1997;92:57–60.
Goldstein NS, Bhanot P. Paucicellular and asymptomatic lymphocytic colitis: expanding the clinicopathologic spectrum of lymphocytic colitis. Am J Clin Pathol. 2004;122:405–11.
Fernandez-Banares F, Casalots J, Salas A, Esteve M, Rosinach M, Forne M, et al. Paucicellular lymphocytic colitis: is it a minor form of lymphocytic colitis? A clinical pathological and immunological study. Am J Gastroenterol. 2009;104:1189–98.
Lamps LW, Lazenby AJ. Colonic epithelial lymphocytosis and lymphocytic colitis: descriptive histopathology versus distinct clinicopathologic entities. Adv Anat Pathol. 2000;7:210–3.
Wang N, Dumot JA, Achkar E, Easley KA, Petras RE, Goldblum JR. Colonic epithelial lymphocytosis without a thickened subepithelial collagen table: a clinicopathologic study of 40 cases supporting a heterogeneous entity. Am J Surg Pathol. 1999;23:1068–74.
Carmack SW, Lash RH, Gulizia JM, Genta RM. Lymphocytic disorders of the gastrointestinal tract: a review for the practicing pathologist. Adv Anat Pathol. 2009;16:290–306.
Bryant DA, Mintz ED, Puhr ND, Griffin PM, Petras RE. Colonic epithelial lymphocytosis associated with an epidemic of chronic diarrhea. Am J Surg Pathol. 1996;20:1102–9.
Yan BM, Shaffer EA. Primary eosinophilic disorders of the gastrointestinal tract. Gut. 2009;58:721–32.
Rothenberg ME. Eosinophilic gastrointestinal disorders (EGID). J Allergy Clin Immunol. 2004;113:11–28 (quiz 9).
Talley NJ, Shorter RG, Phillips SF, Zinsmeister AR. Eosinophilic gastroenteritis: a clinicopathological study of patients with disease of the mucosa, muscle layer, and subserosal tissues. Gut. 1990;31:54–8.
Odze RD, Wershil BK, Leichtner AM, Antonioli DA. Allergic colitis in infants. J Pediatr. 1995;126:163–70.
Machida HM, Catto Smith AG, Gall DG, Trevenen C, Scott RB. Allergic colitis in infancy: clinical and pathologic aspects. J Pediatr Gastroenterol Nutr. 1994;19:22–6.
Clouse RE, Alpers DH, Hockenbery DM, DeSchryver-Kecskemeti K. Pericrypt eosinophilic enterocolitis and chronic diarrhea. Gastroenterology. 1992;103:168–76.
Jakate S, Demeo M, John R, Tobin M, Keshavarzian A. Mastocytic enterocolitis: increased mucosal mast cells in chronic intractable diarrhea. Arch Pathol Lab Med. 2006;130:362–7.
Ogilvie-McDaniel C, Blaiss M, Osborn FD, Carpenter J. Mastocytic enterocolitis: a newly described mast cell entity. Ann Allergy Asthma Immunol. 2008;101:645–6.
Ramsay DB, Stephen S, Borum M, Voltaggio L, Doman DB. Mast cells in gastrointestinal disease. Gastroenterol Hepatol (NY). 2010;6:772–7.
Raithel M, Winterkamp S, Pacurar A, Ulrich P, Hochberger J, Hahn EG. Release of mast cell tryptase from human colorectal mucosa in inflammatory bowel disease. Scand J Gastroenterol. 2001;36:174–9.
Schwab D, Raithel M, Hahn EG. Evidence for mast cell activation in collagenous colitis. Inflamm Res. 1998;47(Suppl 1):S64–5.
Wingren U, Hallert C, Norrby K, Enerback L. Histamine and mucosal mast cells in gluten enteropathy. Agents Actions. 1986;18:266–8.
Blirando K, Milliat F, Martelly I, Sabourin JC, Benderitter M, Francois A. Mast cells are an essential component of human radiation proctitis and contribute to experimental colorectal damage in mice. Am J Pathol. 2011;178:640–51.
Hahn HP, Hornick JL. Immunoreactivity for CD25 in gastrointestinal mucosal mast cells is specific for systemic mastocytosis. Am J Surg Pathol. 2007;31:1669–76.
Pardanani A. Systemic mastocytosis in adults: 2011 update on diagnosis, risk stratification, and management. Am J Hematol. 2011;86:362–71.
Feuerstadt P, Brandt LJ. Colon ischemia: recent insights and advances. Curr Gastroenterol Rep. 2010;12:383–90.
Higgins PD, Davis KJ, Laine L. Systematic review: the epidemiology of ischaemic colitis. Aliment Pharmacol Ther. 2004;19:729–38.
MacDonald PH. Ischaemic colitis. Best Pract Res Clin Gastroenterol. 2002;16:51–61.
Parks DA, Granger DN. Contributions of ischemia and reperfusion to mucosal lesion formation. Am J Physiol. 1986;250:G749–53.
Wheeldon NM, Grundman MJ. Ischaemic colitis as a complication of colonoscopy. BMJ. 1990;301:1080–1.
Carlson RM, Madoff RD. Is “ischemic” colitis ischemic? Dis Colon Rectum. 2011;54:370–3.
Theodoropoulou A, Koutroubakis IE. Ischemic colitis: clinical practice in diagnosis and treatment. World J Gastroenterol. 2008;14:7302–8.
Brandt LJ, Feuerstadt P, Blaszka MC. Anatomic patterns, patient characteristics, and clinical outcomes in ischemic colitis: a study of 313 cases supported by histology. Am J Gastroenterol. 2010;105:2245–52 (quiz 53).
Sotiriadis J, Brandt LJ, Behin DS, Southern WN. Ischemic colitis has a worse prognosis when isolated to the right side of the colon. Am J Gastroenterol. 2007;102:2247–52.
Flobert C, Cellier C, Berger A, Ngo A, Cuillerier E, Landi B, et al. Right colonic involvement is associated with severe forms of ischemic colitis and occurs frequently in patients with chronic renal failure requiring hemodialysis. Am J Gastroenterol. 2000;95:195–8.
Zou X, Cao J, Yao Y, Liu W, Chen L. Endoscopic findings and clinicopathologic characteristics of ischemic colitis: a report of 85 cases. Dig Dis Sci. 2009;54:2009–15.
Scowcroft CW, Sanowski RA, Kozarek RA. Colonoscopy in ischemic colitis. Gastrointest Endosc. 1981;27:156–61.
Fenoglio-Preiser C, Noffsinger A, Stemmermann GN, Lantz PE, Isaacson P. Gastrointestinal pathology. An atlas and text. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2008.
Ye H, Losada M, West AB. Diverticulosis coli: update on a “Western” disease. Adv Anat Pathol. 2005;12:74–80.
Makapugay LM, Dean PJ. Diverticular disease-associated chronic colitis. Am J Surg Pathol. 1996;20:94–102.
Goldstein NS, Leon-Armin C, Mani A. Crohn’s colitis-like changes in sigmoid diverticulitis specimens is usually an idiosyncratic inflammatory response to the diverticulosis rather than Crohn’s colitis. Am J Surg Pathol. 2000;24:668–75.
Brian West A. The pathology of diverticulosis: classical concepts and mucosal changes in diverticula. J Clin Gastroenterol. 2006;40 Suppl 3:S126–31.
Jani N, Finkelstein S, Blumberg D, Regueiro M. Segmental colitis associated with diverticulosis. Dig Dis Sci. 2002;47:1175–81.
Robert ME, Skacel M, Ullman T, Bernstein CN, Easley K, Goldblum JR. Patterns of colonic involvement at initial presentation in ulcerative colitis: a retrospective study of 46 newly diagnosed cases. Am J Clin Pathol. 2004;122:94–9.
Ortega AE, Klipfel N, Kelso R, Petrone P, Roman I, Diaz A, et al. Changing concepts in the pathogenesis, evaluation, and management of solitary rectal ulcer syndrome. Am Surg. 2008;74:967–72.
Tjandra JJ, Fazio VW, Church JM, Lavery IC, Oakley JR, Milsom JW. Clinical conundrum of solitary rectal ulcer. Dis Colon Rectum. 1992;35:227–34.
Martin CJ, Parks TG, Biggart JD. Solitary rectal ulcer syndrome in Northern Ireland. 1971–1980. Br J Surg. 1981;68:744–7.
Chiang JM, Changchien CR, Chen JR. Solitary rectal ulcer syndrome: an endoscopic and histological presentation and literature review. Int J Colorectal Dis. 2006;21:348–56.
Morio O, Meurette G, Desfourneaux V, D’Halluin PN, Bretagne JF, Siproudhis L. Anorectal physiology in solitary ulcer syndrome: a case-matched series. Dis Colon Rectum. 2005;48:1917–22.
Womack NR, Williams NS, Mist JH, Morrison JF. Anorectal function in the solitary rectal ulcer syndrome. Dis Colon Rectum. 1987;30:319–23.
Saul SH, Sollenberger LC. Solitary rectal ulcer syndrome. Its clinical and pathological underdiagnosis. Am J Surg Pathol. 1985;9:411–21.
Vaizey CJ, van den Bogaerde JB, Emmanuel AV, Talbot IC, Nicholls RJ, Kamm MA. Solitary rectal ulcer syndrome. Br J Surg. 1998;85:1617–23.
Warren BF, Dankwa EK, Davies JD. ‘Diamond-shaped’ crypts and mucosal elastin: helpful diagnostic features in biopsies of rectal prolapse. Histopathology. 1990;17:129–34.
Franzin G, Scarpa A, Dina R, Novelli P. “Transitional” and hyperplastic-metaplastic mucosa occurring in solitary ulcer of the rectum. Histopathology. 1981;5:527–33.
Gore S, Shepherd NA, Wilkinson SP. Endoscopic crescentic fold disease of the sigmoid colon: the clinical and histopathological spectrum of a distinctive endoscopic appearance. Int J Colorectal Dis. 1992;7:76–81.
du Boulay CE, Fairbrother J, Isaacson PG. Mucosal prolapse syndrome—a unifying concept for solitary ulcer syndrome and related disorders. J Clin Pathol. 1983;36:1264–8.
Singh B, Mortensen NJ, Warren BF. Histopathological mimicry in mucosal prolapse. Histopathology. 2007;50:97–102.
Ng WK, Chan KW. Postirradiation colitis cystica profunda. Case report and literature review. Arch Pathol Lab Med. 1995;119:1170–3.
Magidson JG, Lewin KJ. Diffuse colitis cystica profunda. Report of a case. Am J Surg Pathol. 1981;5:393–9.
Herman AH, Nabseth DC. Colitis cystica profunda: localized, segmental, and diffuse. Arch Surg. 1973;106:337–41.
Guest CB, Reznick RK. Colitis cystica profunda. Review of the literature. Dis Colon Rectum. 1989;32:983–8.
Koreishi A, Lauwers GY, Misdraji J. Pneumatosis intestinalis: a challenging biopsy diagnosis. Am J Surg Pathol. 2007;31:1469–75.
Heng Y, Schuffler MD, Haggitt RC, Rohrmann CA. Pneumatosis intestinalis: a review. Am J Gastroenterol. 1995;90:1747–58.
Pieterse AS, Leong AS, Rowland R. The mucosal changes and pathogenesis of pneumatosis cystoides intestinalis. Hum Pathol. 1985;16:683–8.
Boerner RM, Fried DB, Warshauer DM, Isaacs K. Pneumatosis intestinalis. Two case reports and a retrospective review of the literature from 1985 to 1995. Dig Dis Sci. 1996;41:2272–85.
Nagata S, Ueda N, Yoshida Y, Matsuda H. Pneumatosis coli complicated with intussusception in an adult: report of a case. Surg Today. 2010;40:460–4.
Lawrentschuk N, Nguyen H. Pneumatosis coli in a case of caecal volvulus. Hosp Med. 2002;63:436–7.
Duron VP, Rutigliano S, Machan JT, Dupuy DE, Mazzaglia PJ. Computed tomographic diagnosis of pneumatosis intestinalis: clinical measures predictive of the need for surgical intervention. Arch Surg. 2011;146:506–10.
Hokama A, Kinjo F, Fujita J. Pneumatosis cystoides intestinalis: radiographic and endoscopic features. Clin Gastroenterol Hepatol. 2009;7:A32.
Suarez V, Chesner IM, Price AB, Newman J. Pneumatosis cystoides intestinalis. Histological mucosal changes mimicking inflammatory bowel disease. Arch Pathol Lab Med. 1989;113:898–901.
Edwards CM, George B, Warren B. Diversion colitis—new light through old windows. Histopathology. 1999;34:1–5.
Roe AM, Warren BF, Brodribb AJ, Brown C. Diversion colitis and involution of the defunctioned anorectum. Gut. 1993;34:382–5.
Neut C, Guillemot F, Colombel JF. Nitrate-reducing bacteria in diversion colitis: a clue to inflammation? Dig Dis Sci. 1997;42:2577–80.
Thorsen AJ. Noninfectious colitides: collagenous colitis, lymphocytic colitis, diversion colitis, and chemically induced colitis. Clin Colon Rectal Surg. 2007;20:47–57.
Geraghty JM, Talbot IC. Diversion colitis: histological features in the colon and rectum after defunctioning colostomy. Gut. 1991;32:1020–3.
Warren BF, Shepherd NA. Diversion proctocolitis. Histopathology. 1992;21:91–3.
Asplund S, Gramlich T, Fazio V, Petras R. Histologic changes in defunctioned rectums in patients with inflammatory bowel disease: a clinicopathologic study of 82 patients with long-term follow-up. Dis Colon Rectum. 2002;45:1206–13.
Griffiths AP, Dixon MF. Microcarcinoids and diversion colitis in a colon defunctioned for 18 years. Report of a case. Dis Colon Rectum. 1992;35:685–8.
Lee KS, Medline A, Shockey S. Indeterminate colitis in the spectrum of inflammatory bowel disease. Arch Pathol Lab Med. 1979;103:173–6.
Bismar MM, Sinicrope FA. Radiation enteritis. Curr Gastroenterol Rep. 2002;4:361–5.
Nielsen OH, Vainer B, Rask-Madsen J. Non-IBD and noninfectious colitis. Nat Clin Pract Gastroenterol Hepatol. 2008;5:28–39.
Leupin N, Curschmann J, Kranzbuhler H, Maurer CA, Laissue JA, Mazzucchelli L. Acute radiation colitis in patients treated with short-term preoperative radiotherapy for rectal cancer. Am J Surg Pathol. 2002;26:498–504.
Gilinsky NH, Burns DG, Barbezat GO, Levin W, Myers HS, Marks IN. The natural history of radiation-induced proctosigmoiditis: an analysis of 88 patients. Q J Med. 1983;52:40–53.
Perrakis N, Athanassiou E, Vamvakopoulou D, Kyriazi M, Kappos H, Vamvakopoulos NC, et al. Practical approaches to effective management of intestinal radiation injury: benefit of resectional surgery. World J Gastroenterol. 2011;17:4013–6.
Hasleton PS, Carr N, Schofield PF. Vascular changes in radiation bowel disease. Histopathology. 1985;9:517–34.
Gelfand MD, Tepper M, Katz LA, Binder HJ, Yesner R, Floch MH. Acute irradiation proctitis in man: development of eosinophilic crypt abscesses. Gastroenterology. 1968;54:401–11.
Sedgwick DM, Howard GC, Ferguson A. Pathogenesis of acute radiation injury to the rectum. A prospective study in patients. Int J Colorectal Dis. 1994;9:23–30.
Haboubi NY, Kaftan SM, Schofield PF. Radiation colitis is another mimic of chronic inflammatory bowel disease. J Clin Pathol. 1992;45:272.
Greenson JK, Stern RA, Carpenter SL, Barnett JL. The clinical significance of focal active colitis. Hum Pathol. 1997;28:729–33.
Shetty S, Anjarwalla SM, Gupta J, Foy CJ, Shaw IS, Valori RM, et al. Focal active colitis: a prospective study of clinicopathological correlations in 90 patients. Histopathology. 2011;59:850–6.
Xin W, Brown PI, Greenson JK. The clinical significance of focal active colitis in pediatric patients. Am J Surg Pathol. 2003;27:1134–8.
Volk EE, Shapiro BD, Easley KA, Goldblum JR. The clinical significance of a biopsy-based diagnosis of focal active colitis: a clinicopathologic study of 31 cases. Mod Pathol. 1998;11:789–94.
Driman DK, Preiksaitis HG. Colorectal inflammation and increased cell proliferation associated with oral sodium phosphate bowel preparation solution. Hum Pathol. 1998;29:972–8.
Goldstein NS, Cinenza AN. The histopathology of nonsteroidal anti-inflammatory drug-associated colitis. Am J Clin Pathol. 1998;110:622–8.
Schneider EN, Havens JM, Scott MA, Goldblum JR, Greenson JK, Shaffer RA, et al. Molecular diagnosis of Campylobacter jejuni infection in cases of focal active colitis. Am J Surg Pathol. 2006;30:782–5.
Lee FD. Drug-related pathological lesions of the intestinal tract. Histopathology. 1994;25:303–8.
Wong NA, Penman ID, Campbell S, Lessells AM. Microscopic focal cryptitis associated with sodium phosphate bowel preparation. Histopathology. 2000;36:476–8.
Conflict of interest
The authors hereby confirm that neither the manuscript nor any part of it has been published or is being considered for publication elsewhere. We declare that there are no potential sources of financial conflicts, including stock ownership and patents.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Khor, T.S., Fujita, H., Nagata, K. et al. Biopsy interpretation of colonic biopsies when inflammatory bowel disease is excluded. J Gastroenterol 47, 226–248 (2012). https://doi.org/10.1007/s00535-012-0539-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-012-0539-6