
Abstract
The pathogenesis of all immune-mediated inflammatory diseases has been carefully studied over the past several decades, but it is only recently that we have come to appreciate common pathways and genes. This is especially true for the inflammatory bowel diseases (IBD) Crohn’s disease and ulcerative colitis, where a keener appreciation of the contributions of genetics, environment, and immune response have been dissected. In fact, in many ways, IBD has become the model for studying such disorders. The complex nature of interactions is continuing to be defined, and novel therapies targeting defects in these interactions have been developed and are being tested in the clinic. The era of bench to bedside has finally matured, and cures for debilitating diseases are now in sight. This review describes our current state of knowledge of each component of IBD pathogenesis. What has evolved is a clearer picture and novel targets for therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Significant advances in the understanding of disease pathogenesis have led to a therapeutic revolution in inflammatory bowel disease (IBD). Importantly, a number of different factors have contributed to this unprecedented leap in our knowledge base (animal models, direct analysis of human diseased tissue, identification of disease susceptibility genes, and the recognition of the role of environmental factors). The advances have come from a variety of sources: geneticists, microbiologists, epidemiologists, and mucosal immunologists reflecting the important input of various components to disease pathogenesis. These have contributed to the identification of major pathways and therapeutic targets and to the development of novel disease-modifying drugs. The results of the clinical studies represent, for the first time, a true proof of concept approach, where one can ask whether pathways or molecules identified via animal models or human tissues are truly important in the disease process in vivo.
When one looks at the disease process it can be divided into three phases—initiation, augmentation, and perpetuation/regulation. Both the innate and adaptive immune system contribute to the first two phases, while defects in regulation reflect alterations in adaptive immunity (Table 1). Agents that target the late phases of the initiation process (T-cell activation) and all phases of immunoregulation have a high likelihood of controlling disease activity. When one evaluates a target’s potential for efficacy, one needs to characterize whether it is able to prevent a cell from getting to the site of inflammation, prevent the cell’s activation once it gets there, or prevent the effects of cellular activation if the other two processes fail. Most therapies have been directed at the third component, since it is easier to target products of the immune system (anti-cytokines, anti-cytokine receptors). The results of these interventions have added further insights into disease pathogenesis (i.e., does intervention work or not, and if not, why not?).
Like all other immune-mediated inflammatory diseases (IMIDs), the actual development of disease reflects the contributions of host genetics, an environmental trigger, and the consequent immune response. Crohn’s disease has been the poster child for the study of complex genetic disorders, being the first disease in which a replicated genetic mutation was identified (NOD2/CARD15) [1, 2]. This success was followed by the identification of a number of additional genetic associations, including OCTN1/2 [3], MDR1α [4], and most recently, ATG16L [5, 6] and IL23R [7]. Interestingly many of these genes encode molecules involved in innate immunity (see below) or epithelial barrier function (defective function). This is particularly important because the majority of the current biologic agents target effector molecules of the adaptive and not innate immune system or the epithelial barrier. Thus, these genetic links not only provide new clues to pathogenesis but also suggest novel therapeutic targets.
To understand these targets one needs to have a basic understanding of disease mechanisms. This is not an easy task as hypotheses of disease pathogenesis change and evolve. There are, however, basic concepts that have remained constant. It is clear that after we account for a genetic predisposition and environmental trigger, the culprit responsible for mediating the disease process is an altered immune response [8, 9]. In the normal intestine the immunologic tone is one of tolerance or suppression of immune responses against non-pathogens in the gut lumen. These non-pathogens include both commensal flora and dietary antigens. In IBD there is growing evidence that this tolerant state, at least to specific components of the flora, is lost (the presence of ANCA, ASCA, OmpC, and CBir serum antibodies is evidence for loss of tolerance) [10, 11]. Tolerance is mediated by cells that regulate immune responses (regulatory cells) [12]. These may be ineffectively activated or maintained in IBD [13–15]. Hence, in the absence of regulation, an inflammatory response elicited by a (probably harmless) trigger is able to persist, resulting in an aggressive inflammatory reaction. While cellular constituents are responsible for the inflammation, it is more the products of the cells and the effects that they have on other local cell populations that give rise to what we see as the actual disease. Depending on the type of disease (Crohn’s disease versus ulcerative colitis [UC]), the inflammatory response may be quite different, and even within Crohn’s disease there may be multiple distinct pathways operating in different patients (the simplest example is fistulizing vs. inflammatory vs. fibrostenotic Crohn’s [16]). Furthermore, there is increasing evidence that the inflammatory mediators may change with time, either as a result of therapy (anti-tumor necrosis factor [TNF]) or as part of the natural course of disease [17, 18]. This would suggest that a patient might lose response to a given agent with time, requiring them to move out of class for the next therapy. With these possibilities in mind let us look at the pathogenesis of IBD in its current state.
Current paradigms for disease pathogenesis
Genetics
As mentioned above, similar to all immune-mediated inflammatory diseases, the development of IBD reflects the interaction of at least three components; a genetic predisposition, an environmental trigger, and an unregulated or dysregulated immune response. Given the enormous advances in technology and understanding of the human genome, new genetic associations for IBD (more so for Crohn’s disease) are being identified monthly. What we have learned is that Crohn’s disease is more of a genetically regulated disease than UC [19–21], that environmental factors remain key (e.g., smoking) [22], that these diseases are multigenic likely accounting for disease heterogeneity [23], and that many of the genes identified to date support defects in innate immunity and epithelial barrier function [1–3, 5–7]. Just having a mutation in one susceptibility gene (e.g., CARD15/NOD2) is not a sine qua non for the development of disease. Indeed, in some populations the carriage rate in healthy controls for CARD15 mutations associated with Crohn’s disease is 20% [24]. Thus, one cannot use the findings of a gene mutation for the purposes of genetic counseling or disease predictability. Interestingly, and as would be expected, having these mutations is associated with younger age of onset of disease and a more aggressive disease phenotype [25, 26] (Table 2).
The same holds for some of the newer gene associations described; specifically the IL23R mutations and the ATG16L [6] mutation. The former is involved in the IL12/23 pathway of inflammation thought to be important in the pathogenesis of Crohn’s disease (see below). The latter is associated with control of a process called autophagy, where cells under stress initiate intracellular programs that result in cell death. This process is thought to prevent the inflammation associated with necrotic cell death, and while we do not fully understand the functional consequence of the ATG16L gene mutation, failure to stimulate autophagy may promote inflammation [6].
Lastly, the defect in NOD2/CARD15 or OCTNI/II may affect the host’s ability to localize and eradicate bacteria that gain entry to the host [3]. Failure to eliminate such triggers could result in aberrant inflammatory responses and, possibly more importantly, allow for persistence of antigens that are capable of triggering an adaptive immune response.
Environmental factors (initiation)
As alluded to above, it has become quite clear from genetic studies that genetics alone is insufficient to explain the development of IBD. Monozygotic twin studies show the concordance rate for disease being 40–60% [19, 20, 27, 28]. Since this number is not 100%, the environment must play a key role in the development of disease. Several triggers have been identified and there is strong evidence that no single factor or agent is responsible for the development of disease (e.g., Mycobacterium paratuberculosis [29]). Clinical observation has shown that agents that break the mucosal barrier are common triggers of disease. These include nonsteroidal anti-inflammatory drugs (NSAIDs), antibiotics, and viral and bacterial infections. While smoking is a widely replicated risk factor for the development of Crohn’s disease, it has a protective effect for UC. The basis for this difference is unclear. Lastly, while diet and stress are not clearly linked to the development of IBD, there is a growing appreciation of the fact that diet alters flora or that components of the diet (e.g., metals) can alter the functional properties of the flora. Stress reactions have been shown to alter immune responses but the lack of reproducible quantitative data has left this potential trigger largely unexplored [30]. Importantly, different patient subpopulations express different genetic mutations. It is appealing to speculate that different environmental factors trigger disease in these different patient populations. Thus, “Crohn’s disease type 1” may be the result of a bacterial infection in a patient with a NOD2/CARD15 mutation, while “Crohn’s disease type 2” results from the effect of smoking in a patient with an ATG16L variant, etc. Understanding these connections will enable appropriate counseling in specific patient subpopulations (Table 3).
Immune response (augmentation)
There are two basic components to the immune response; the innate and adaptive response. Innate immunity is the more primitive form of immunity that lacks specificity and memory for pathogens that it has seen. It is responsible for the early initial immune response where localization and eradication of invading pathogens is critical to the host’s survival [30, 31]. Adaptive immunity evolved because of the inability of the innate immune system to augment responses upon reinfection and the fact that larger pathogen loads easily overwhelm the innate response. Adaptive immunity is characterized by specificity and memory and is largely mediated by lymphocytes, T and B cells, that express antigen receptors on their surface [32]. Cytokines produced by T cells in response to infection orchestrate an organized directed immune response that not only results in the eradication of an infectious agent but also gives rise to memory cells that prevent disease upon re-exposure to the offending microbe.
Innate immunity
While the innate response was largely ignored for years because of its limited repertoire and lack of specificity, newer studies have strongly supported the fact that defects in innate immunity have significant consequences to the host and can alter adaptive immunity as well [33]. In fact, the innate immune response is largely responsible for paving the way for the adaptive response. In the intestine, innate immunity includes the epithelial barrier and phagocytic cells within the lamina propria (macrophages, dendritic cells [DCs], and neutrophils). Interestingly, patients exhibiting genetic defects in innate immunity (e.g., chronic granulomatous disease, Hermansky Pudlak syndrome) [34, 35] have an increased incidence of IBD. This correlation has led to the development of agents used to boost innate immunity (e.g., granulocyte macrophage colony-stimulating factor [GM-CSF]) as therapeutic agents in IBD [36].
Adaptive immunity
Despite the growing appreciation for the role of innate immunity in regulating immune responses in the gut, the adaptive immune system, specifically T lymphocytes, has been most tightly correlated with disease pathogenesis [37, 38]. T cells become activated, secrete cytokines and affect all other cell types within a local environment (macrophages, DCs, neutrophils, epithelium, endothelial cells, stromal elements). Both human and murine studies have led to the recognition that different T-cell populations are aberrantly activated in Crohn’s disease versus UC [39, 40] and that even within Crohn’s disease there may be distant subsets that drive the inflammatory response. Lastly, as alluded to above, there may be multiple pathways interacting simultaneously or changes in pathways as the disease evolves.
Th1 versus Th2 versus Th17
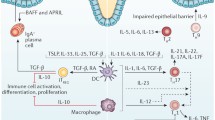
Much to the chagrin of gastroenterologists, immunologists feel most comfortable categorizing populations of T cells as capable of secreting specific profiles of cytokines. The list of cytokines grows every year and many exhibit overlapping functions. What must be kept in mind is the fact that the immune response is plastic, changing constantly with environmental cues and that modest adjustments in either the timing, amount, or composition of cytokines in the microenvironment can have profound effects on the visible immune response or immunopathology. This may be why some cytokines that were thought to play key roles in IBD pathogenesis were shown to be less relevant when targeted in human trials. Successful neutralization with specific agents (e.g., fontilizumab—anti-interferon [IFN]γ) had limited effects on disease activity [41] (Fig. 1).

The pathogenesis of Crohn’s disease relates to a genetic predisposition, an environmental trigger and an aberrant immune response. This latter response reflects defects in both innate and adaptive immunity. Innate immune defects account for altered responses to commensal flora including the production of pro-inflammatory cytokines IL12 or IL23 resulting in either IFNγ/TNFα or IL17 production by T cells. These cytokines act on tissue macrophages to release tissue-altering enzymes that account for the disease expression. LT Lymphotoxin
Th1-mediated immune responses are typically evoked in response to an intracellular pathogen (e.g., virus, mycobacteria, histoplasma). The coordinated immune response elicited is meant to localize the infectious agent (e.g., form a granuloma) and secrete factors that either promote intracellular killing (IFNγ, TNFα) or induce the differentiation of cells that mediate killing (cytotoxic T lymphocytes). The hallmark of a Th1 response is the granuloma seen in purified protein derivative (PPD) skin responses (delayed type hypersensitivity reaction) or the granulomas seen in either mycobacterial infections (caseating) or Crohn’s disease or sarcoid (non-caseating) [42]. T-cell activation occurs in the setting of an antigen presented by an antigen-presenting cell (APC; DC, macrophage, B cell). In the presence of interleukin (IL) I2 secreted by the APC, the T cell is now programmed to produce pro-inflammatory cytokines: IFNγ, TNFα, and IL2 (under the control of a master transcriptional regulator Tbet [43]). These cytokines act on local cell populations to promote intracellular killing (superoxides, peroxides), enhance recruitment of other inflammatory cells (TNF increases vascular adhesion molecules such as MadCAM the target for α4β7-expressing lymphocytes [44]), enhance secretion of chemoattractant cytokines, (chemokines), and promote local tissue destruction (induction of the secretion of collagenases, elastases, matrix metalloproteinases [45]). In the setting of an infectious disease, this response is meant to localize the infection and make the local microenvironment inhospitable for the pathogen. In the setting of a Th1-driven immune-mediated disease, the activation of this pathway is aberrant. IL12, composed of two chains—p40 and p35, is part of a larger family of diverse cytokines [46, 47]. IL12p40 is also used by another pro-inflammatory cytokine, IL23 (p40/p19 heterodimer) that affects a different T-cell population (Th17, see below). Blocking IL12 inhibits the generation of a Th1 response and blocks the production of the downstream cytokines (T-cell derived TNFα, IFNγ, and IL2) although other cell types (natural killer [NK] cells and macrophages) can produce TNFα and IFNγ and are not governed by IL12. Inhibition of IL12 is not without potential consequence. IL12 receptor and IL12-deficient families have been described and are prone to mycobacterial infections [48, 49].
Recently an additional Th subset has been described, Th17 cells [50]. These cells produce IL17 and IL22, both pro-inflammatory cytokines capable of promoting local tissue destruction. TH17 cells are activated by the combination of IL6 and transforming growth factor (TGF) β (stimulating the transcription factor RORγt [51]) and are induced to survive and further differentiate to mature IL17-secreting cells by IL23. As mentioned above, IL23 shares the p40 subunit with IL12, so antibody against p40 results in the inhibition of both IL12 and IL23. IL17 and IL22 are found at increased levels in inflamed Crohn’s mucosa suggesting that these cytokines may play a role in disease pathogenesis [52, 53]. This has clearly been shown to be the case in some animal models where selective inhibition of IL23 and not IL12 prevents mucosal inflammation. This shift in paradigm from Th1 to ThIL17 is supported by the lack of effect of anti-IFNγ in the treatment of Crohn’s.
Another Th subset is the TH2 cell. These cells secrete IL4, IL5, and IL13 [54]. TH2 cells promote atopy with induction of IgE responses and eosinophil and mast cell activation. Initially, UC was thought to be a TH2-cell-mediated disease, but the absence of IL4 in UC tissues and the observation that both IL13 and IFNγ are found at elevated levels in UC mucosa changed the dogma [55]. Recently one group [39] has suggested that the IL13 found in UC tissues comes from a non-T cell, an NK T cell, and that IL13 targets the epithelial cell to become dysfunctional [56]. Thus, UC may be more of a superficial epithelial injury disorder, while Crohn’s disease may be more of a submucosal disease reflecting immune activation deeper in the tissue. The fact that infliximab has some salutary effect in UC further suggests that the aberrant immune response in this disease is more of a mixed picture [57] (Fig. 2).

The pathogenesis of ulcerative colitis relates to a genetic predisposition, an environmental trigger and an aberrant immune response. In contrast to the changes seen in CD, a different T cell subset is activated in UC. This subset produces IL13 as well as IFNγ resulting in epithelial dysfunction, antibody production, and immune complex formation resulting in complement activation and mast cell degranulation. The effects of these activating events account for distinct pathologic findings and specific disease expression
Having made the point that there are specific T-cell subpopulations, the evidence for the sovereignty of these cells in humans is less clear-cut. Rarely is only one cell type activated in an immune response and there is always plasticity in the response. For example, there are reports in humans of T cells producing both IL17 and IFNγ [58], as well as others producing, IL10 and IFNγ [59]. Equally as important, under certain conditions and microenvironmental cues, different effector cells may be recruited to the inflamed tissue. For example, CCR9+ CD4+ T cells are attracted to the inflamed small intestinal epithelium expressing the chemokine CCL25 (TECK) [60], while CXCR3+ memory T cells migrate towards inflamed colonic epithelium expressing IFN-γ-inducible chemokines [61]. The classifications of T-cell subpopulations on the basis of cytokine secretion profiles are therefore helpful but not absolute. Furthermore, the findings described above underscore the point that an unregulated immune response of any type is poorly tolerated in the gastrointestinal (GI) tract.
Amplification of the immune response
If the trigger/initiation of the inflammatory disorder is activation of the T cell, the amplification process reflects the downstream effects of this activation. T-cell derived cytokines act on cells in the local environment to produce chemokines that attract additional inflammatory cells into the local environment. These cells are regulated differently than cells that have grown up in the suppressed environment of the gut. They may be more easily activated and induced to secrete pro-inflammatory factors and tissue destructive enzymes. Since the variety of chemokines is great and redundancy overwhelming, inhibiting one chemokine is not likely to have a significant effect on the inflammation in IBD. A global chemokine inhibitor may, however, prove to be more effective.
Amplification is furthered by the enhancement of the expression of mucosal adhesion molecules (e.g., MadCAM1) [44] on the endothelium of vessels in the gut. Enhanced expression of addressins promotes the egress of cells from the peripheral blood. These too are less vulnerable to the rules and regulations guiding the activation of lymphocytes that are native inhabitants of the gut. Agents that block entry of cells into the gut would be predicted to have a beneficial long-term effect on inflammation. Since mucosal T cells from patients with IBD stay alive longer than normal mucosal T cells [62] and do not traffic to other sites, anti-adhesion molecule antibodies would not be expected to have an immediate effect on mucosal inflammation. In contrast, they would be expected to have a beneficial effect on maintaining remission. This appears to be the case in the clinical trials of anti-adhesion molecules performed to date [63].
Perpetuation and regulation
Regulatory cells
The last and probably most critical pathway in the gut mucosa is the one involved in immune regulation. The immunologic tone of the GI tract, in contrast to the systemic immune system, is one of suppression. Normal individuals generally do not generate systemic immune responses against commensal flora or dietary antigens. Regulatory cells are responsible for the immunologically suppressed tone in the gut mucosa. Regulatory cells come in many flavors (T cells, B cells, DCs, macrophages, NK T cells), but the cell type that has received the greatest attention is the regulatory T cell [64, 65]. These too come in many varieties and are abundant and possibly redundant in the GI tract. Various CD4+ Tregs have been described as being involved in suppressing mucosal responses. Tr1 cells secrete IL10 [66], a potent immunosuppressive cytokine that may be responsible for suppressing responses to commensal flora. IL10-deficient mice develop a Crohn’s-like inflammatory disease [67]. TH3 cells produce TGFβ, another potent immunosuppressant cytokine that promotes IgA production while suppressing T- and B-cell activation [68]. One of the most popular regulatory T cells is the CD4+25+ Treg [64, 65]. These are more systemic regulatory cells that require the transcription factor FoxP3 for their activation. The absence of FoxP3 in humans (IPEX syndrome) [69, 70] and mouse (Scurfy) results in autoimmune endocrine disease, immunodeficiency, and an autoimmune enteropathy (not colitis but small bowel pathology in a small subset of patients). FoxP3+ T cells are present in normal to higher numbers in IBD patients, suggesting that these diseases are not a result of defects in this population. Lastly, CD8+ T cells are regulatory in the gut as well. A number of different CD8+ Tregs have been described. Some of these (TrE cells) [15, 71] have been shown to be lacking in patients with IBD and may therefore be relevant to disease pathogenesis (Fig. 3).

Regulatory T cells are thought to be responsible for the controlled physiologic inflammation seen in the normal state. Several different Tregs exist in the GI tract including IL10 secreting CD4+ T cells (Tr1). TGFβ secreting T cells (TH3) and the contact dependent type, CD4+ CD25+ FoxP3+ Tregs
A popular concept is that defects in Tregs allow for the perpetuation of active inflammation. While there are no direct studies supporting this concept, papers such as those cited above, as well as a documented defect in oral tolerance, in both IBD patients and a significant number of first-degree relatives, support this hypothesis [72, 73]. Thus, therapies directed at increasing the activation of Tregs would be of value in disease therapy.
Tertiary inflammatory pathways
What has been focused on above relates to the activation and dysregulation of the immune response in IBD. However, what we actually see as the disease is the result of downstream events. These events are multiple and redundant and include tissue-altering enzymes such as collagenase, elastase, matrix metalloproteinases, superoxides, hydrogen peroxide, leukotrienes, and prostaglandins. Inhibiting any of these pathways can have a salutary effect on the inflammatory process, but if the upstream stimulus is not controlled, alternative tertiary inflammatory pathways can be activated and inflammation persists.
From the various sections described above it is clear that we have come to a point where we are able to dissect disease pathways (Fig. 4). Understanding each and every component of the inflammatory response has clearly aided our efforts in developing new approaches to disease management. Whether these translate into useful therapeutic tools can be determined only in direct human clinical trials.

Model of IBD pathogenesis identifying the key elements (genetics, environment and immunity) along with subtle alterations that can occur in response to modifiers (cigarettes, metals, diet, etc.)
References
Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–6.
Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603.
Peltekova VD, Wintle RF, Rubin LA, Amos CI, Huang Q, Gu X, et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat Genet. 2004;36:471–5.
Brant SR, Panhuysen CI, Nicolae D, Reddy DM, Bonen DK, Karaliukas R, et al. MDR1 Ala893 polymorphism is associated with inflammatory bowel disease. Am J Hum Genet. 2003;73:1282–92.
Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–11.
Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604.
Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3.
Podolsky DK. Lessons from genetic models of inflammatory bowel disease. Acta Gastroenterol Belg. 1997;60:163–5.
Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117:514–21.
Braun J, Targan SR. Multiparameter analysis of immunogenetic mechanisms in clinical diagnosis and management of inflammatory bowel disease. Adv Exp Med Biol. 2006;579:209–18.
Duchmann R, Kaiser I, Hermann E, Mayet W, Ewe K, Meyer zum Buschenfelde KH. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease (IBD). Clin Exp Immunol. 1995;102:448–55.
Mowat AM, Viney JL. The anatomical basis of intestinal immunity. Immunol Rev. 1997;156:145–66.
Maul J, Loddenkemper C, Mundt P, Berg E, Giese T, Stallmach A, et al. Peripheral and intestinal regulatory CD4+ CD25(high) T cells in inflammatory bowel disease. Gastroenterology. 2005;128:1868–78.
Mottet C, Uhlig HH, Powrie F. Cutting edge: cure of colitis by CD4+ CD25+ regulatory T cells. J Immunol. 2003;170:3939–43.
Brimnes J, Allez M, Dotan I, Shao L, Nakazawa A, Mayer L. Defects in CD8+ regulatory T cells in the lamina propria of patients with inflammatory bowel disease. J Immunol. 2005;174:5814–22.
Sachar DB. Genomics and phenomics in Crohn’s disease. Gastroenterology. 2002;122:1161–2.
Spencer DM, Veldman GM, Banerjee S, Willis J, Levine AD. Distinct inflammatory mechanisms mediate early versus late colitis in mice. Gastroenterology. 2002;122:94–105.
Rutgeerts P, Van Assche G, Vermeire S. Optimizing anti-TNF treatment in inflammatory bowel disease. Gastroenterology. 2004;126:1593–610.
Halme L, Paavola-Sakki P, Turunen U, Lappalainen M, Farkkila M, Kontula K. Family and twin studies in inflammatory bowel disease. World J Gastroenterol. 2006;12:3668–72.
Orholm M, Binder V, Sorensen TI, Rasmussen LP, Kyvik KO. Concordance of inflammatory bowel disease among Danish twins. Results of a nationwide study. Scand J Gastroenterol. 2000;35:1075–81.
Satsangi J, Parkes M, Jewell DP, Bell JI. Genetics of inflammatory bowel disease. Clin Sci (Lond). 1998;94:473–8.
Bernstein CN, Rawsthorne P, Cheang M, Blanchard JF. A population-based case control study of potential risk factors for IBD. Am J Gastroenterol. 2006;101:993–1002.
Brant SR, Shugart YY. Inflammatory bowel disease gene hunting by linkage analysis: rationale, methodology, and present status of the field. Inflamm Bowel Dis. 2004;10:300–11.
Esters N, Pierik M, van Steen K, Vermeire S, Claessens G, Joossens S, et al. Transmission of CARD15 (NOD2) variants within families of patients with inflammatory bowel disease. Am J Gastroenterol. 2004;99:299–305.
Weiss B, Shamir R, Bujanover Y, Waterman M, Hartman C, Fradkin A, et al. NOD2/CARD15 mutation analysis and genotype-phenotype correlation in Jewish pediatric patients compared with adults with Crohn’s disease. J Pediatr. 2004;145:208–12.
Abreu MT, Taylor KD, Lin YC, Hang T, Gaiennie J, Landers CJ, et al. Mutations in NOD2 are associated with fibrostenosing disease in patients with Crohn’s disease. Gastroenterology. 2002;123:679–88.
Halfvarson J, Bodin L, Tysk C, Lindberg E, Jarnerot G. Inflammatory bowel disease in a Swedish twin cohort: a long-term follow-up of concordance and clinical characteristics. Gastroenterology. 2003;124:1767–73.
Halfvarson J, Jess T, Bodin L, Jarnerot G, Munkholm P, Binder V, et al. Longitudinal concordance for clinical characteristics in a Swedish–Danish twin population with inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:1536–44.
Chiba M, Fukushima T, Horie Y, Iizuka M, Masamune O. No Mycobacterium paratuberculosis detected in intestinal tissue, including Peyer’s patches and lymph follicles, of Crohn’s disease. J Gastroenterol. 1998;33:482–7.
Drossman DA. Presidential address: gastrointestinal illness and the biopsychosocial model. Psychosom Med. 1998;60:258–67.
Medzhitov R, Janeway C Jr. Innate immune recognition: mechanisms and pathways. Immunol Rev. 2000;173:89–97.
Janeway CA Jr, Bottomly K. Signals and signs for lymphocyte responses. Cell. 1994;76:275–85.
Medzhitov R, Janeway CA Jr. Innate immune recognition and control of adaptive immune responses. Semin Immunol. 1998;10:351–3.
Huang JS, Noack D, Rae J, Ellis BA, Newbury R, Pong AL, et al. Chronic granulomatous disease caused by a deficiency in p47(phox) mimicking Crohn’s disease. Clin Gastroenterol Hepatol. 2004;2:690–5.
Schinella RA, Greco MA, Cobert BL, Denmark LW, Cox RP. Hermansky–Pudlak syndrome with granulomatous colitis. Ann Int Med. 1980;92:20–3.
Dieckgraefe BK, Korzenik JR. Treatment of active Crohn’s disease with recombinant human granulocyte–macrophage colony-stimulating factor. Lancet. 2002;360:1478–80.
Kugathasan S, Saubermann LJ, Smith L, Kou D, Itoh J, Binion DG, et al. Mucosal T-cell immunoregulation varies in early and late inflammatory bowel disease. Gut. 2007;56:1696–705.
Brown SJ, Mayer L. The immune response in inflammatory bowel disease. Am J Gastroenterol. 2007;102:2058–69.
Fuss IJ, Heller F, Boirivant M, Leon F, Yoshida M, Fichtner-Feigl S, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113:1490–7.
Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA, et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–70.
van Assche G. Emerging drugs to treat Crohn’s disease. Expert Opin Emerg Drugs. 2007;12:49–59.
Kobayashi K, Kaneda K, Kasama T. Immunopathogenesis of delayed-type hypersensitivity. Microsc Res Tech. 2001;53:241–5.
Glimcher LH. Trawling for treasure: tales of T-bet. Nat Immunol. 2007;8:448–50.
Berlin C, Berg EL, Briskin MJ, Andrew DP, Kilshaw PJ, Holzmann B, et al. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell. 1993;74:185–95.
MacDonald TT, Bajaj-Elliott M, Pender SL. T cells orchestrate intestinal mucosal shape and integrity. Immunol Today. 1999;20:505–10.
Alber G, Al-Robaiy S, Kleinschek M, Knauer J, Krumbholz P, Richter J, Schoeneberger S, Schuetze N, Schulz S, Toepfer K, Voigtlaender R, Lehmann J, Mueller U. Induction of immunity and inflammation by interleukin-12 family members. Ernst Schering Res Found Workshop 2006:107–27.
Hunter CA. New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat Rev Immunol. 2005;5:521–31.
Elloumi-Zghal H, Barbouche MR, Chemli J, Bejaoui M, Harbi A, Snoussi N, et al. Clinical and genetic heterogeneity of inherited autosomal recessive susceptibility to disseminated Mycobacterium bovis bacille calmette-guerin infection. J Infect Dis. 2002;185:1468–75.
Sakai T, Matsuoka M, Aoki M, Nosaka K, Mitsuya H. Missense mutation of the interleukin-12 receptor beta1 chain-encoding gene is associated with impaired immunity against Mycobacterium avium complex infection. Blood. 2001;97:2688–94.
Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–52.
Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–74.
Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70.
Fuss IJ, Becker C, Yang Z, Groden C, Hornung RL, Heller F, et al. Both IL-12p70 and IL-23 are synthesized during active Crohn’s disease and are down-regulated by treatment with anti-IL-12 p40 monoclonal antibody. Inflamm Bowel Dis. 2006;12:9–15.
Umetsu DT, DeKruyff RH. Th1 and Th2 CD4+ cells in the pathogenesis of allergic diseases. Proc Soc Exp Biol Med. 1997;215:11–20.
Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol. 2003;3:521–33.
Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–64.
Rutgeerts P, Sandborn WJ, Feagan BG, Reinisch W, Olson A, Johanns J, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353:2462–76.
Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–61.
Denning TL, Qi H, Konig R, Scott KG, Naganuma M, Ernst PB. CD4+ Th cells resembling regulatory T cells that inhibit chronic colitis differentiate in the absence of interactions between CD4 and class II MHC. J Immunol. 2003;171:2279–86.
Hosoe N, Miura S, Watanabe C, Tsuzuki Y, Hokari R, Oyama T, et al. Demonstration of functional role of TECK/CCL25 in T lymphocyte–endothelium interaction in inflamed and uninflamed intestinal mucosa. Am J Physiol Gastrointest Liver Physiol. 2004;286:G458–66.
Lu B, Humbles A, Bota D, Gerard C, Moser B, Soler D, et al. Structure and function of the murine chemokine receptor CXCR3. Eur J Immunol. 1999;29:3804–12.
Neurath MF, Finotto S, Fuss I, Boirivant M, Galle PR, Strober W. Regulation of T-cell apoptosis in inflammatory bowel disease: to die or not to die, that is the mucosal question. Trends Immunol. 2001;22:21–6.
Ghosh S, Goldin E, Gordon FH, Malchow HA, Rask-Madsen J, Rutgeerts P, et al. Natalizumab for active Crohn’s disease. N Engl J Med. 2003;348:24–32.
Stephens GL, Shevach EM. Foxp3+ regulatory T cells: selfishness under scrutiny. Immunity. 2007;27:417–9.
Zheng Y, Rudensky AY. Foxp3 in control of the regulatory T cell lineage. Nat Immunol. 2007;8:457–62.
Cong Y, Weaver CT, Lazenby A, Elson CO. Bacterial-reactive T regulatory cells inhibit pathogenic immune responses to the enteric flora. J Immunol. 2002;169:6112–9.
Rennick DM, Fort MM, Davidson NJ. Studies with IL-10-/- mice: an overview. J Leukoc Biol. 1997;61:389–96.
Coffman RL, Lebman DA, Shrader B. Transforming growth factor beta specifically enhances IgA production by lipopolysaccharide-stimulated murine B lymphocytes. J Exp Med. 1989;170:1039–44.
Ruemmele FM, Brousse N, Goulet O. Autoimmune enteropathy: molecular concepts. Curr Opin Gastroenterol. 2004;20:587–91.
Ochs HD, Gambineri E, Torgerson TR. IPEX, FOXP3 and regulatory T-cells: a model for autoimmunity. Immunol Res. 2007;38:112–21.
Allez M, Brimnes J, Dotan I, Mayer L. Expansion of CD8+ T cells with regulatory function after interaction with intestinal epithelial cells. Gastroenterology. 2002;123:1516–26.
Kraus TA, Toy L, Chan L, Childs J, Cheifetz A, Mayer L. Failure to induce oral tolerance in Crohn’s and ulcerative colitis patients: possible genetic risk. Ann N Y Acad Sci. 2004;1029:225–38.
Kraus TA, Toy L, Chan L, Childs J, Mayer L. Failure to induce oral tolerance to a soluble protein in patients with inflammatory bowel disease. Gastroenterology. 2004;126:1771–8.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mayer, L. Evolving paradigms in the pathogenesis of IBD. J Gastroenterol 45, 9–16 (2010). https://doi.org/10.1007/s00535-009-0138-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-009-0138-3