
Abstract
Biliary atresia (BA) is a progressive fibro-obliterative cholangiopathy affecting the extra- and intrahepatic biliary tree to various degrees and resulting in obstructive bile flow, cholestasis and icterus in neonates. It is the most common cause of pediatric liver transplantation. The etiology of BA is still unclear, although there is some evidence pointing to viral, toxic, and multiple genetic factors. For new therapeutic options other than liver transplantation to be developed, a greater understanding of the pathogenesis of BA is indispensable. The fact that the pathology of BA develops during a period of biliary growth and remodeling suggests an involvement of developmental anomalies. Recent studies indicate an association of the etiology of BA with some genetic factors such as laterality genes, epigenetic regulation and/or microRNA function. In this paper, we present an overview of recent advances in the understanding of the disease focusing on bile duct developmental anomaly.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Biliary atresia (BA) is a progressive fibro-obliterative cholangiopathy affecting the extra- and intrahepatic biliary tree to various degrees and resulting in obstructive bile flow, cholestasis and icterus in neonates [1–3]. Without treatment such as the Kasai procedure, a hepatoportoenterostomy performed to establish bile drainage from microscopic residual bile ductules within the liver, progressive hepatic fibrosis leads to cirrhosis, portal hypertension, liver failure and death by the age of 2 years [1, 2]. It is a rare disease in the Western world, with an incidence of 1:15000–19000 live births [3], whereas in Asia the incidence is clearly higher, for example, ranging from 1:5400 to 5800 live births in Taiwan [4] and 1:9000–10000 live births in Japan. BA is classified based on the level of extrahepatic obstruction of the biliary tree [1]. In type 1 BA (about 5 %), the biliary duct is obstructed at the level of the common bile duct. In type 2 BA (about 2 %), obstruction occurs at the level of the common hepatic duct. In type 3 BA (>90 %), the most proximal part of the extrahepatic biliary duct in the hepatic portal region is obstructed and fibrotic without any macroscopic remnants of the hepatic ducts. Additional cystic dilatation of the extrahepatic bile duct remnants may be present. Even in type 3 BA, microscopic residual bile ductules of variable size remain in continuity with the intrahepatic biliary tree. BA can also be separated into two clinical entities, namely an embryonic or fetal (congenital) form and a postnatal progressive (posteriori) form. Some patients with the congenital form of BA also present with a constellation of anomalies known as biliary atresia splenic malformation (BASM) syndrome. These patients (10–20 % in the Western world) are more often female, and they exhibit variable combinations of associated malformations including asplenia/polysplenia, abnormal abdominal situs, intestinal malrotation, abdominal vascular anomaly, congenital heart disease and/or pancreatic malformations. In the remaining 80 % of cases, BA occurs in isolation without any associated disorders.
In this paper, we describe recent advances in the study of the etiology of BA, especially focusing on understanding it as a developmental anomaly.
Abnormal developmental theory for BA
The pathogenesis of BA is unknown. It is likely to be multifactorial, in that environmental and genetic factors may interact in the process of its development into an obstructive cholangiopathy [1, 5]. Viral infection, autoimmunity, inflammation, abnormal development, and genetics have all been proposed as potentially involved [6–8]. A theory proposed by Mack [9] suggests that BA is caused by a specific perinatal hepatobiliary viral infection. In this view, immune-mediated damage in response to infection leads to the eventual obliteration of the bile ducts. As an alternative explanation, BA is thought to result from developmental anomaly, lead to the leakage of bile and consequent inflammation.
During human embryogenesis, the transverse septum takes on a ventral position where an endodermal projection of the ventral portion of the primitive gut (the hepatic diverticulum) gradually develops and gives rise to the liver. From the caudal segment of the hepatic diverticulum, cells differentiate to form the gallbladder and the cystic, hepatic, and common bile ducts. Adjacent endoderm cells undergo pancreatic specification and form the endocrine and exocrine pancreas [10]. Bile drains from the hepatocyte-lined bile canaliculi into intrahepatic bile tracts through a transitional region called the Canal of Hering. Both hepatocytes and cholangiocytes are derived from hepatoblasts. Several steps of differentiation and morphogenesis are required for normal intrahepatic bile duct development. First, periportal hepatoblasts are induced to differentiate into cholangiocyte precursors, forming ring structures known as ductal plates around the portal vein branches. At discrete locations within the ductal plates, tubular structures known as primitive ductal structures (PDS) form; these eventually give rise to bile ducts. Subsequently, precursors that fail to be incorporated into the bile ducts regress, leaving mature ducts in place [11–13]. While extrahepatic bile ducts are more severely affected, BA involves both the extrahepatic and intrahepatic biliary systems. The abnormal development explanation for BA centers on the concept that developmental errors occur somewhere in the differentiation and morphogenesis of the biliary system.
Genetic factors for biliary atresia
Since the congenital form may be associated with ductal plate malformations characterized by an abnormal persistence of ductal plate-like structures, it is suggested that a mutation in the genes regulating bile duct development could act as a susceptibility factor or a modifier gene. In one genetic study that examined 102 cases of BA, nine patients harbored a missense mutation in jagged 1, a Notch signaling ligand [14]. Another study investigated the expression of Notch receptors in BA livers and found that notch 3 expression was increased in neovessels and mesenchymal cells [15]. These results suggest that abnormal Notch signaling may predispose to BA. It is important to note that about 10 % of BA patients also have additional congenital defects related to left–right axis abnormalities, suggesting that proteins regulating left–right patterning may be involved in the disease. These include the cilium-associated protein Inversin, the Nodal co-factor Cryptic (CFC1) and the zinc finger transcription factor ZIC3. For example, mice with a deletion of the invs gene exhibit BA-like disease [16]. However, anomalies of biliary system were not found in humans with INVS mutations [17]. A heterozygous mutation in CFC1 was found in a patient with BA and polysplenia [18], and a CFC1 polymorphism (Ala145Thr) was identified in 5 out of 10 patients, twice the frequency observed in control patients [19]. This alteration in CFC1 is not sufficient to induce BA, however: BA occurred in only one of two brothers harboring the same amino acid substitution [20]. Two patients with BA and congenital heart defects had mutations in ZIC3 [21]. Moreover, a comparative analysis of genes expressed in livers exhibiting the congenital and posteriori forms of BA revealed a unique pattern of gene expression in the congenital form. A number of chromatin-modifying genes and imprinted genes were upregulated, but, most significantly, the expression of the laterality genes SPROUTY4, LEFTYA and ZIC3 differed between the congenital and posteriori forms of BA [22]. An analysis of genes in livers exhibiting posteriori forms of BA revealed anomalies in the expression of several genes involved in morphogenesis, fibrogenesis, transcriptional regulation and cell signaling [23]. However, whether these gene anomalies indicate pathogenic mechanisms or result from the progression of the disease remains uncertain at the moment.
Animal models of extrahepatic biliary duct anomalies
Although the extrahepatic and intrahepatic biliary systems are ultimately in direct contact, their origins are distinct: lineage tracing studies have shown that the extrahepatic biliary system and the ventral pancreas may share common progenitor cells that are distinct from those that form the liver. It has been demonstrated using a Pdx1 transcription factor-cre mouse strain that both the extrahepatic biliary system and the ventral pancreas are derived from Pdx1-positive cells, while the liver and intrahepatic bile ducts are not [24].
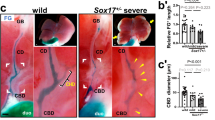
The Sox17 transcription factor, a protein involved in the formation of endodermal organs, controls specification of the liver and bile ducts. On approximately embryonic day (E) 8.5 in mice, cells in the ventral foregut express both Pdx1 and Sox17. Subsequently, the Sox17+/Pdx1+ domain becomes divided into a Sox17+ biliary primordium and a Pdx1+ pancreatic primordium. Inactivation of Sox17 in the ventral foregut causes the loss of induction into extrahepatic biliary ducts and leads to the formation of ectopic pancreatic tissue, while misexpression of Sox17 in the Pdx1+ domain suppresses pancreatic development and induces a ductal epithelial fate, suggesting that Sox17 regulates the extrahepatic biliary duct/pancreatic cell fate decision [24].
The appropriate differentiation of the extrahepatic biliary duct and ventral pancreatic lineages is also regulated by Hes1, a transcriptional effector of Notch signaling. In Hes1-deficient mice, the extrahepatic biliary duct differentiates towards a pancreatic phenotype, suggesting that Hes1 represses the differentiation of extrahepatic biliary ducts toward a pancreatic fate [25, 26]. This phenotype is similar to that of Sox17 conditional knock-out mice, suggesting an interaction between Hes1 and Sox17 during extrahepatic biliary duct development. Indeed, inactivation of Sox17 results in a down-regulation of Hes1 expression, while inactivation of Hes1 leads to a paradoxical expansion of the Sox17-domain [24, 27], suggesting the existence of a Sox17–Hes1 feedback loop.
Other factors required for appropriate extrahepatic biliary duct development include HNF6, HNF1β, and Hhex. The transcription factors HNF6 and HNF1β were the first to be identified as transcriptional regulators of biliary development [11, 28]. In the absence of either factor the ducts develop as cystic structures of variable size, and at birth there is an abnormal persistence of ductal plate-like structures, a feature typical of ductal plate malformations described in human biliary diseases. HNF6 can bind to the Hnf1b gene, suggesting that the two factors are linked in a gene cascade regulating biliary differentiation and morphogenesis. Ablation of Hhex at the hepatic diverticulum stage results in hypoplastic liver with loss of hepatoblast marker expression. Around E16.5–E18.5, these livers start to express hepatocyte/hepatoblast markers but also develop numerous cysts which are lined with a mixed population of epithelial cells displaying biliary and hepatobiliary phenotypes [29]. Misexpression of Sox17 results in an upregulation of HNF6, HNF1β and Hhex expression [24], suggesting that these transcription factors are downstream effectors of Sox17. Given the extrahepatic biliary duct anomalies seen in mice deficient in Hnf6, Hnf1β, FoxF1, Hes1, Pdx1 and Hhex, these genes may be involved in the etiology of extrahepatic biliary cysts, but no genetic evidence presently supports this hypothesis [30].
MicroRNAs and BA pathogenesis
MicroRNAs (miRNAs) are small noncoding RNAs that bind to target mRNAs and induce mRNA cleavage, destabilization or translational repression. Although miRNAs are involved in several diseases of the gastro-intestinal tract [31], information on the function of miRNAs in liver development remains insufficient. Inhibition of miRNA synthesis by liver-specific knockout of Dicer, which is required for the processing of all miRNAs, using albumin-cre or Alfp-cre mice strains, results in postnatal liver defects [32, 33]. In these experiments, however, Cre-mediated recombination of the Dicer locus did not occur early enough to allow analysis of miRNA function in the prenatal period. On the other hand, a genome-wide study identified a set of 38 miRNAs whose expression in liver changes significantly during the process of liver development (from late gestation to the perinatal period) [34]. Among these 38 miRNAs, miR-30a and miR-30c are found to be expressed specifically in cholangiocytes. Additionally, removing miR-30a in zebrafish caused defects in bile duct formation, suggesting that miR-30a is required for biliary development. Potential targets of miR-30a include the epidermal growth factor (EGF) receptor, a regulator of cholangiocyte proliferation, as well as Activin A, a secreted factor that belongs to the TGFβ family and may control biliary differentiation. MiR-15a, a repressor of cdc25a, also represses proliferation of cholangiocytes [35].
Recently, it was reported that miR-29a/29b-1 are upregulated in the Rhesus rotavirus (RRV)-BALB/c model of BA. Consistent with this increase in miR-29 levels, levels of DNA methyltransferase 3, a direct target of miR-29, are likewise decreased [36]. The reciprocal relation between miR-29 and methyltransferase gene expression in experimental BA is remarkable given the recent observation that DNA hypomethylation leads to bile defects in a zebrafish model and correlates with clinical BA [36, 37]. We also found that, among 12 miRNAs highly expressed in a normal intrahepatic biliary epithelial cell line (HIBEpiC) [38], five of them, including miR-29b, exhibited specifically increased expression in BA livers compared with normal livers and those with other metabolic diseases (unpublished data). However, recent reports suggest that miR-29 is also involved in the synthesis of collagen type I in liver fibrosis [39, 40], suggesting that miR-29 was upregulated as a result of the progress of fibrosis resulting from BA.
On the other hand, recent studies have identified stable populations of miRNAs present in cell-free plasma and serum preparations [41–44]. As these circulating miRNAs can have altered expressions or be released from injured and diseased tissues, their concentrations can be indicative of certain related disorders [45–47]. In BA, it has been demonstrated that the miR-200b/429 cluster is significantly increased in the sera of BA patients, suggesting that circulating miR-200b/429 levels are diagnostic values for BA [48], although the elevation of miR-200b/429 in BA sera may result from the destruction of the extrahepatic bile duct, and the exact relation between miR-200b/429 and BA pathogenesis is unknown.
DNA modification and BA pathogenesis
DNA methylation is the only genetically programmed DNA modification process in mammals that is involved in the regulation of several biological processes, including gene transcription, X-chromosome inactivation, genomic imprinting and chromatin modification [49–51]. Methylation of cytosine at CpG residues leads to repression of gene expression [52], and changes in DNA methylation can be elicited by drugs, toxins, viruses [53] and genetic defects [54]. It has been demonstrated that DNA methylation is significantly reduced in bile duct cells from BA patients compared to patients with other infantile cholestatic disorders. Intrahepatic biliary defects and upregulated hepatic expression of IFN-γ pathway genes are caused by genetic or pharmacological inhibition of DNA methylation in zebrafish, suggesting a possible etiologic link between decreased DNA methylation, activation of IFN-γ signaling, and biliary defects in patients [37]. In addition, defective maintenance of DNA methylation may lead to the development of many autoimmune diseases, including systemic lupus erythematosus (SLE), rheumatoid arthritis and multiple sclerosis [55–57]. It was demonstrated that the expression of CD11a is significantly decreased in CD4+ T cells derived from BA patients. The hypermethylation of promoter regulatory elements contributes to the lower CD11a expression in CD4+ T cells of infants with BA, and this abnormal expression of CD11a may contribute to the pathogenesis of BA [58]. These results suggest the importance of abnormal DNA methylation in the development of BA disorders.
Genome-wide association studies for BA
Genome-wide association studies have identified BA susceptibility loci on several chromosomes [59, 60]. Leyva-Vega et al. [60] identified two unrelated BA patients with overlapping heterozygous deletions of 2q37.3. Patient 1 had a 1.76 Mb (280 SNP) heterozygous deletion containing 30 genes. Patient 2 had a 5.87 Mb (1346 SNP) heterozygous deletion containing 55 genes. This suggests that the overlapping 1.76 Mb deletion on chromosome 2q37.3 from 240936900 to 242692820 contains the critical region and that the genes within this region could be candidates for conferring susceptibility to BA. On the other hand, Garcia-Barcelo et al. [59] identified a susceptibility locus for BA on 10q24.2. They showed that the likelihood of developing BA is influenced by DNA variants in a region spanning 129 kb and encompassing the XPNPEP1 and ADD3 genes. These studies indicate that the identification of putative BA susceptibility loci not only opens new fields of investigation into the mechanisms underlying BA but may also provide new clues for the development of preventive and curative strategies [59].
Conclusion
Although the etiology of BA is unknown, developmental anomalies have been proposed as an explanation for its pathophysiology. It is suggested that some proteins regulating left–right patterning may be involved in BA pathogenesis, though these gene anomalies do not appear to be solely responsible for the pathogenic mechanisms. Studies carried out in twins have demonstrated that non-genetic factors also play an important role in mediating BA pathogenesis [61], but the identities of these remain obscure, as no common environmental factor that triggers disease progression has yet been identified. Recent studies indicate that epigenetic factors such as DNA methylation or novel regulatory factors such as miRNAs may be involved in BA pathogenesis. Genome-wide association studies using next-generation sequencing technology may provide novel insights into BA pathogenesis in the future. In addition, studies of iPS cells derived from BA patients may reveal abnormal differentiation into cholangiocytes in BA.
BA remains the most serious liver disease in children. Even using the best available surgical techniques to correct BA, the outcome and prognosis are still inadequate. A better understanding of the etiology and pathogenesis of BA is needed in order to develop new strategies for early diagnosis, treatment and prevention, as well as the avoidance of surgery-related complications.
References
Hartley JL, Davenport M, Kelly DA. Biliary atresia. Lancet. 2009;374(9702):1704–13.
Bassett MD, Murray KF. Biliary atresia: recent progress. J Clin Gastroenterol. 2008;42(6):720–9.
Pakarinen MP, Rintala RJ. Surgery of biliary atresia. Scand J Surg. 2011;100(1):49–53.
Hsiao CH, Chang MH, Chen HL, Lee HC, Wu TC, Lin CC, et al. Universal screening for biliary atresia using an infant stool color card in Taiwan. Hepatology. 2008;47(4):1233–40.
Muraji T, Suskind DL, Irie N. Biliary atresia: a new immunological insight into etiopathogenesis. Expert Rev Gastroenterol Hepatol. 2009;3(6):599–606.
Sokol RJ, Mack C. Etiopathogenesis of biliary atresia. Semin Liver Dis. 2001;21(4):517–24.
Tan CE, Driver M, Howard ER, Moscoso GJ. Extrahepatic biliary atresia: a first-trimester event? Clues from light microscopy and immunohistochemistry. J Pediatr Surg. 1994;29(6):808–14.
Arikan C, Berdeli A, Ozgenc F, Tumgor G, Yagci RV, Aydogdu S. Positive association of macrophage migration inhibitory factor gene-173G/C polymorphism with biliary atresia. J Pediatr Gastroenterol Nutr. 2006;42(1):77–82.
Mack CL. The pathogenesis of biliary atresia: evidence for a virus-induced autoimmune disease. Semin Liver Dis. 2007;27(3):233–42.
Santos JL, Carvalho E, Bezerra JA. Advances in biliary atresia: from patient care to research. Braz J Med Biol Res. 2010;43(6):522–7.
Clotman F, Lannoy VJ, Reber M, Cereghini S, Cassiman D, Jacquemin P, et al. The onecut transcription factor HNF6 is required for normal development of the biliary tract. Development. 2002;129(8):1819–28.
Lemaigre FP. Development of the biliary tract. Mech Dev. 2003;120(1):81–7.
Shiojiri N, Katayama H. Secondary joining of the bile ducts during the hepatogenesis of the mouse embryo. Anat Embryol (Berl). 1987;177(2):153–63.
Kohsaka T, Yuan ZR, Guo SX, Tagawa M, Nakamura A, Nakano M, et al. The significance of human jagged 1 mutations detected in severe cases of extrahepatic biliary atresia. Hepatology. 2002;36(4 Pt 1):904–12.
Flynn DM, Nijjar S, Hubscher SG, de Goyet Jde V, Kelly DA, Strain AJ, et al. The role of Notch receptor expression in bile duct development and disease. J Pathol. 2004;204(1):55–64.
Mazziotti MV, Willis LK, Heuckeroth RO, LaRegina MC, Swanson PE, Overbeek PA, et al. Anomalous development of the hepatobiliary system in the Inv mouse. Hepatology. 1999;30(2):372–8.
Schon P, Tsuchiya K, Lenoir D, Mochizuki T, Guichard C, Takai S, et al. Identification, genomic organization, chromosomal mapping and mutation analysis of the human INV gene, the ortholog of a murine gene implicated in left–right axis development and biliary atresia. Hum Genet. 2002;110(2):157–65.
Bamford RN, Roessler E, Burdine RD, Saplakoglu U, dela Cruz J, Splitt M, et al. Loss-of-function mutations in the EGF-CFC gene CFC1 are associated with human left–right laterality defects. Nat Genet. 2000;26(3):365–9.
Davit-Spraul A, Baussan C, Hermeziu B, Bernard O, Jacquemin E. CFC1 gene involvement in biliary atresia with polysplenia syndrome. J Pediatr Gastroenterol Nutr. 2008;46(1):111–2.
Jacquemin E, Cresteil D, Raynaud N, Hadchouel M. CFCI gene mutation and biliary atresia with polysplenia syndrome. J Pediatr Gastroenterol Nutr. 2002;34(3):326–7.
Ware SM, Peng J, Zhu L, Fernbach S, Colicos S, Casey B, et al. Identification and functional analysis of ZIC3 mutations in heterotaxy and related congenital heart defects. Am J Hum Genet. 2004;74(1):93–105.
Zhang DY, Sabla G, Shivakumar P, Tiao G, Sokol RJ, Mack C, et al. Coordinate expression of regulatory genes differentiates embryonic and perinatal forms of biliary atresia. Hepatology. 2004;39(4):954–62.
Chen L, Goryachev A, Sun J, Kim P, Zhang H, Phillips MJ, et al. Altered expression of genes involved in hepatic morphogenesis and fibrogenesis are identified by cDNA microarray analysis in biliary atresia. Hepatology. 2003;38(3):567–76.
Spence JR, Lange AW, Lin SC, Kaestner KH, Lowy AM, Kim I, et al. Sox17 regulates organ lineage segregation of ventral foregut progenitor cells. Dev Cell. 2009;17(1):62–74.
Fukuda A, Kawaguchi Y, Furuyama K, Kodama S, Horiguchi M, Kuhara T, et al. Ectopic pancreas formation in Hes1-knockout mice reveals plasticity of endodermal progenitors of the gut, bile duct, and pancreas. J Clin Invest. 2006;116(6):1484–93.
Sumazaki R, Shiojiri N, Isoyama S, Masu M, Keino-Masu K, Osawa M, et al. Conversion of biliary system to pancreatic tissue in Hes1-deficient mice. Nat Genet. 2004;36(1):83–7.
Zaret KS, Grompe M. Generation and regeneration of cells of the liver and pancreas. Science. 2008;322(5907):1490–4.
Coffinier C, Gresh L, Fiette L, Tronche F, Schutz G, Babinet C, et al. Bile system morphogenesis defects and liver dysfunction upon targeted deletion of HNF1beta. Development. 2002;129(8):1829–38.
Hunter MP, Wilson CM, Jiang X, Cong R, Vasavada H, Kaestner KH, et al. The homeobox gene Hhex is essential for proper hepatoblast differentiation and bile duct morphogenesis. Dev Biol. 2007;308(2):355–67.
Raynaud P, Carpentier R, Antoniou A, Lemaigre FP. Biliary differentiation and bile duct morphogenesis in development and disease. Int J Biochem Cell Biol. 2011;43(2):245–56.
O’Hara SP, Mott JL, Splinter PL, Gores GJ, LaRusso NF. MicroRNAs: key modulators of posttranscriptional gene expression. Gastroenterology. 2009;136(1):17–25.
Hand NJ, Master ZR, Le Lay J, Friedman JR. Hepatic function is preserved in the absence of mature microRNAs. Hepatology. 2009;49(2):618–26.
Sekine S, Ogawa R, Ito R, Hiraoka N, McManus MT, Kanai Y, et al. Disruption of Dicer1 induces dysregulated fetal gene expression and promotes hepatocarcinogenesis. Gastroenterology. 2009;136(7):2304–15. (e1–e4).
Hand NJ, Master ZR, Eauclaire SF, Weinblatt DE, Matthews RP, Friedman JR. The microRNA-30 family is required for vertebrate hepatobiliary development. Gastroenterology. 2009;136(3):1081–90.
Lee SO, Masyuk T, Splinter P, Banales JM, Masyuk A, Stroope A, et al. MicroRNA15a modulates expression of the cell-cycle regulator Cdc25A and affects hepatic cystogenesis in a rat model of polycystic kidney disease. J Clin Invest. 2008;118(11):3714–24.
Hand NJ, Horner AM, Master ZR, Boateng LA, LeGuen C, Uvaydova M, et al. MicroRNA profiling identifies miR-29 as a regulator of disease-associated pathways in experimental biliary atresia. J Pediatr Gastroenterol Nutr. 2012;54(2):186–92.
Matthews RP, Eauclaire SF, Mugnier M, Lorent K, Cui S, Ross MM, et al. DNA hypomethylation causes bile duct defects in zebrafish and is a distinguishing feature of infantile biliary atresia. Hepatology. 2011;53(3):905–14.
Kawahigashi Y, Mishima T, Mizuguchi Y, Arima Y, Yokomuro S, Kanda T, et al. MicroRNA profiling of human intrahepatic cholangiocarcinoma cell lines reveals biliary epithelial cell-specific microRNAs. J Nihon Med Sch. 2009;76(4):188–97.
Ogawa T, Iizuka M, Sekiya Y, Yoshizato K, Ikeda K, Kawada N. Suppression of type I collagen production by microRNA-29b in cultured human stellate cells. Biochem Biophys Res Commun. 2010;391(1):316–21.
Roderburg C, Urban GW, Bettermann K, Vucur M, Zimmermann H, Schmidt S, et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology. 2011;53(1):209–18.
Lawrie CH, Gal S, Dunlop HM, Pushkaran B, Liggins AP, Pulford K, et al. Detection of elevated levels of tumour-associated microRNAs in serum of patients with diffuse large B-cell lymphoma. Br J Haematol. 2008;141(5):672–5.
Gilad S, Meiri E, Yogev Y, Benjamin S, Lebanony D, Yerushalmi N, et al. Serum microRNAs are promising novel biomarkers. PLoS One. 2008;3(9):e3148.
Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105(30):10513–8.
Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18(10):997–1006.
Morimura R, Komatsu S, Ichikawa D, Takeshita H, Tsujiura M, Nagata H, et al. Novel diagnostic value of circulating miR-18a in plasma of patients with pancreatic cancer. Br J Cancer. 2011;105(11):1733–40.
Laterza OF, Lim L, Garrett-Engele PW, Vlasakova K, Muniappa N, Tanaka WK, et al. Plasma microRNAs as sensitive and specific biomarkers of tissue injury. Clin Chem. 2009;55(11):1977–83.
Zahm AM, Thayu M, Hand NJ, Horner A, Leonard MB, Friedman JR. Circulating microRNA is a biomarker of pediatric Crohn disease. J Pediatr Gastroenterol Nutr. 2011;53(1):26–33.
Zahm AM, Hand NJ, Boateng LA, Friedman JR. Circulating microRNA is a biomarker of biliary atresia. J Pediatr Gastroenterol Nutr. 2012;55(4):366–9.
Geiman TM, Robertson KD. Chromatin remodeling, histone modifications, and DNA methylation-how does it all fit together? J Cell Biochem. 2002;87(2):117–25.
Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2(1):21–32.
Chow J, Heard E. X inactivation and the complexities of silencing a sex chromosome. Curr Opin Cell Biol. 2009;21(3):359–66.
Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31(2):89–97.
Kruger DH, Schroeder C, Santibanez-Koref M, Reuter M. Avoidance of DNA methylation. A virus-encoded methylase inhibitor and evidence for counter selection of methylase recognition sites in viral genomes. Cell Biophys. 1989;15(1–2):87–95.
Glenn CC, Nicholls RD, Robinson WP, Saitoh S, Niikawa N, Schinzel A, et al. Modification of 15q11–q13 DNA methylation imprints in unique Angelman and Prader-Willi patients. Hum Mol Genet. 1993;2(9):1377–82.
Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 1990;33(11):1665–73.
Karouzakis E, Gay RE, Michel BA, Gay S, Neidhart M. DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2009;60(12):3613–22.
Baranzini SE, Mudge J, van Velkinburgh JC, Khankhanian P, Khrebtukova I, Miller NA, et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature. 2010;464(7293):1351–6.
Dong R, Zhao R, Zheng S, Zheng Y, Xiong S, Chu Y. Abnormal DNA methylation of ITGAL (CD11a) in CD4+ T cells from infants with biliary atresia. Biochem Biophys Res Commun. 2012;417(3):986–90.
Garcia-Barcelo MM, Yeung MY, Miao XP, Tang CS, Cheng G, So MT, et al. Genome-wide association study identifies a susceptibility locus for biliary atresia on 10q24.2. Hum Mol Genet. 2010;19(14):2917–25.
Leyva-Vega M, Gerfen J, Thiel BD, Jurkiewicz D, Rand EB, Pawlowska J, et al. Genomic alterations in biliary atresia suggest region of potential disease susceptibility in 2q37.3. Am J Med Genet A. 2010;152A(4):886–95.
Smith BM, Laberge JM, Schreiber R, Weber AM, Blanchard H. Familial biliary atresia in three siblings including twins. J Pediatr Surg. 1991;26(11):1331–3.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Nakamura, K., Tanoue, A. Etiology of biliary atresia as a developmental anomaly: recent advances. J Hepatobiliary Pancreat Sci 20, 459–464 (2013). https://doi.org/10.1007/s00534-013-0604-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00534-013-0604-4