
Abstract
Purpose
Biosimilars are supported by limited clinical data at the time of approval. Recently, Nivestim™, a biosimilar of reference of filgrastim, was approved for prevention of chemotherapy-related febrile neutropenia (FN). To add clinical experience to this new biosimilar, we performed a study to compare the effectiveness of Nivestim™ with reference filgrastim and pegfilgrastim in FN prevention in patients receiving high-risk FN chemotherapy.
Methods
This is a comparative cohort study, with retrospective data collection. Three cohorts were identified according to the type of primary prophylaxis employed over different time periods: reference filgrastim (2004–2006), pegfilgrastim (2007–2008) and biosimilar filgrastim (2011–2012). The study included female patients with early breast cancer that received FN primary prophylaxis during (neo)adjuvant docetaxel/doxorubicin/cyclophosphamide (TAC).
Results
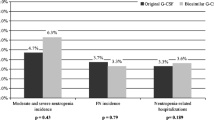
Reference filgrastim cohort included 147 patients and pegfilgrastim and biosimilar filgrastim cohorts 139 and 134 patients, respectively. FN rates per patient/cycle were 16 % (95 % confidence interval (CI) 10.2–22.5 %)/3 % (95 % CI 2.1–4.7 %) in the reference filgrastim group, 9 % (95 % CI 4.5–14.6 %)/2 % (95 % CI 1.3–3.6 %) in the pegfilgrastim group and 16 % (95 % CI 10.0–22.9 %)/4 % (95 % CI 2.5–5.3 %) in the biosimilar filgrastim cohort. The median absolute neutrophil count (ANC) at FN presentation was lower in the biosimilar group in comparison with reference filgrastim. FN episodes with ANC < 100 cells/μL were more frequent in the biosimilar group (50 %) when compared with reference filgrastim (4 %) and pegfilgrastim (6 %). No differences concerning FN complications were seen, with the exception of more chemotherapy delays in the biosimilar group when compared with pegfilgrastim.
Conclusion
No differences in biosimilar effectiveness were detected. The clinical relevance of the profound neutropenia found in the biosimilar cohort needs further attention.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Febrile neutropenia (FN) is a frequent complication of chemotherapy, which carries substantial risk of morbidity and mortality and holds a negative impact on health care use and costs [1–3]. FN-related events are of particular concern in early breast cancer since delays in chemotherapy administration, dose reductions, and premature suspension of treatment impair optimal chemotherapy dose delivery which may compromise the curative intent [4, 5].
TAC (docetaxel/doxorubicin/cyclophosphamide) chemotherapy has been considered one of the standards of care in early breast cancer patients. In the original TAC trial published in 2005, one of the major toxicities reported was FN, affecting 24 % of patients despite prophylactic ciprofloxacin [6]. In subsequent TAC studies, FN rate was reduced by the addition of primary granulocyte colony-stimulating factor (G-CSF) prophylaxis [7, 8]. This strategy is supported by current guidelines which recommend the routine use of G-CSF prophylaxis in regimens with FN risk superior to 20 % [9–11].
Filgrastim (Neupogen™, Amgen) and its pegylated form pegfilgrastim (Neulasta™, Amgen) are two G-CSF formulations widely used in clinical practice associated with substantial costs [12]. In a time of considerable economic constraints, G-CSF biosimilars are an emerging class of biopharmaceutical agents that may become an interesting cost-saving alternative to cope with the increasing burden of cancer. Unlike generic molecules, biosimilars are not identical copies but rather a similar version of the biologic originator product and are considered a change in clinical practice.
Recently, Nivestim™ (Hospira), a biosimilar of Amgen reference filgrastim, received marketing approval for FN prevention by the European Medicines Agency (EMA) and Australian Therapeutic Goods Administration (TGA). The approval was supported by a phase III trial [13] that demonstrated bioequivalence of biosimilar filgrastim in mean duration of severe neutropenia, mean time to recovery of absolute neutrophil count (ANC) and incidence of FN, the EMA recommended endpoints. Even though the bioequivalence requirements were met, this was only one single equivalence study which included a small heterogeneous breast cancer population, both from curative and metastatic settings, receiving a high-risk FN anthracycline-taxane regimen.
To add out of trial clinical experience to this new biosimilar, we performed a study to compare the effectiveness of Nivestim™ with Amgen reference filgrastim and pegfilgrastim in FN prevention in early breast cancer patients receiving TAC regimen.
Methods
Study design and patients
This is a non-interventional comparative cohort study, with retrospective data collection, conducted in a tertiary cancer centre. The study received Institutional Review Board approval. Three cohorts of female patients with early breast cancer who underwent TAC regimen were selected according to the type of G-CSF primary prophylaxis employed over different time periods in our centre: reference filgrastim (2004–2006), pegfilgrastim (2007–2008) and biosimilar filgrastim (Nivestim™) (2011–2012). In order to ensure patients’ consecutive inclusion, cohorts were identified from our pharmacy department database, which maintained a record of TAC cycles administered and G-CSF provided to patients. The current study was a development of a previous work published in 2011 [14] that reported our FN rate in a sample of 147 patients and which now established our 2004–2006 cohort. To obtain approximately the same sample size in subsequent groups, we performed a convenience sampling with no formal statistical hypothesis stated previously. The two latest study periods were thus dictated by changes in G-CSF primary prophylaxis institutional protocols plus the time needed to obtain approximately the same number of patients in each cohort.
The standard TAC protocol consisted of docetaxel 75 mg/m2, doxorubicin 50 mg/m2 and cyclophosphamide 500 mg/m2, all administered intravenous on day 1 every 3 weeks [6, 7]. To be included, patients had to have completed at least one cycle of TAC. All patients had finished treatment at the time of analysis. To estimate TAC dose intensity in each group, we evaluated the relative dose intensity (RDI) [15] in each patient for docetaxel, doxorubicin and cyclophosphamide separately. RDI was calculated as the ratio (%) of the average dose intensity actually delivered per week and the reference dose intensity per week. The average dose intensity was defined as the cumulative dose delivered (mg/m2) divided by the actual duration of chemotherapy exposure (weeks). In our centre, only patients with ECOG 0 to 1 were eligible for TAC regimen. Ciprofloxacin, consisting of 500 mg twice daily on days 5 through 11 of each cycle, was used as a concomitant FN prophylactic measure at clinician’s discretion before 2011 and from 2011 onwards in the first cycle as part of an institutional protocol. Reference and biosimilar filgrastim was available in 300 or 480-μg syringes. The recommended dose is 5 μg/kg daily; nevertheless, doses between 4 and 8 μg/kg/day appear to be also effective [16]. For convenience in our clinical practice, we administer 300 or 480 μg in patients ≤ 75 and >75 kg, respectively. Pegfilgrastim was administered as a 6-mg single injection.
The FN management protocol was the same during the three time periods. If low-risk FN episode, a combination of intravenous amoxicillin/clavulanate and quinolone (or clarithromycin if patient was already on quinolone or in case of quinolone allergy) was started. These patients remained under surveillance in our 24-h emergency department and later were discharged with oral antibiotic if clinically stable. If no clinical improvement occurred, escalation to a broader spectrum antibacterial protocol (mainly piperacillin/tazobactam and aminoglycoside) was performed. Patients with high-risk features (e.g. hypotension or organ dysfunction) at FN diagnosis started broad-spectrum antibiotics upfront. Admission to the ward (‘hospitalization’) was carried out whenever prolonged amoxicillin/clavulanate and quinolone intravenous therapy was necessary or broad-spectrum antibiotics were needed.
Data collection
Data on patient demographic and clinical characteristics and FN episodes were collected from clinical charts and laboratory records. In the few cases where a record of an FN episode managed in other health care facilities existed, we requested information on the event. Information on the TAC cycles, G-CSF dose, schedule and number of administrations was extracted from clinical records and pharmacy department database. In the first 67 patients of the reference filgrastim cohort, a patient telephone interview was done to confirm the G-CSF pattern of use, but given the agreement between clinical charts, pharmacy database and patient information, no further interviews were considered necessary.
Effectiveness endpoints
Our primary endpoint was the proportion of patients and TAC cycles with FN (defined as body temperature ≥ 38 °C concurrent with ANC ≤ 500 cells/μL). Other endpoints analysed were ANC at FN diagnosis, FN complications (hospitalization rate and duration, septic shock defined as FN with fluid refractory hypotension requiring vasopressors, and death), FN-related chemotherapy delays (delay defined as ≥4 days from the planned date), reductions (defined as a ≥15 % reduction in the planned dose), and early termination due to FN.
Statistical analysis
Exact binomial two-sided 95 % confidence intervals were calculated for the proportion of patients and cycles with FN in each G-CSF treatment group. In an exploratory analysis, we performed comparisons between the three G-CSF groups regarding the effectiveness endpoints using a Pearson’s chi-squared test for categorical outcomes (where an asymptotic test was not appropriate, a Pearson’s chi-squared test with simulated p value based on 2000 replicates was used instead) and a Kruskal-Wallis test for the quantitative outcomes. Where applicable, post hoc pairwise comparisons were performed using a Wilcoxon rank sum test with continuity correction and Bonferroni p value adjustment. All tests were two-sided and the considered significance value was 5 %. Statistical analysis was done in R software (http://www.R-project.org).
Results
Demographic and clinical characteristics
Table 1 shows the demographic and clinical baseline characteristics of the three cohorts. The reference filgrastim cohort included 147 patients and pegfilgrastim and biosimilar cohorts 139 and 134 patients, respectively. Characteristics were well balanced across the three cohorts with the exception of age and treatment setting. In the biosimilar cohort, patients were younger (median age of 48 vs. 52 years in the other cohorts) with only 10 % of patients with ≥60 years old. There was also an increase of TAC use in the neoadjuvant setting in pegfilgrastim and biosimilar cohorts.
TAC dose intensity, G-CSF pattern of use and antibacterial prophylaxis
Median RDI was 100 % for each TAC drug in every cohort. In both reference and biosimilar filgrastim groups, the majority of patients started G-CSF prophylaxis 1 to 3 days after chemotherapy (91 and 83 %, respectively) and completed greater than or equal to seven injections of daily G-CSF per cycle (88 and 87 %, respectively) (Table 2). Almost all patients (98 %) in the pegfilgrastim cohort performed the G-CSF injection on the day after chemotherapy.
Ciprofloxacin was used as an additional FN preventive measure to G-CSF in 110/761 (14 %) of TAC cycles in the pegfilgrastim group and in 186/761 (24 %) in the biosimilar group but in none of the cycles in the reference filgrastim cohort. There was an increased use of ciprofloxacin in the first cycle of the biosimilar group (69 vs. 17 % pegfilgrastim).
FN rate and complications
The FN rates per patient/cycle were 16/3 % in the reference filgrastim group, 16/4 % in the biosimilar group and 9/2 % in the pegfilgrastim group (Table 3). In our exploratory analysis, no significant differences were found between the three groups. In all groups, there was clear prevalence for FN episodes to occur in the first cycle (data not shown).
There were differences in ANC at FN diagnosis between the three groups (Table 3). Post hoc analyses revealed that median ANC at FN diagnosis was lower in the biosimilar group in comparison with reference filgrastim (p = 0.015) but not with pegfilgrastim (p = 0.440) or between reference filgrastim and pegfilgrastim (p = 0.223). FN episodes with ANC < 100 cells/μL at diagnosis were more frequent in the biosimilar group when compared with pegfilgrastim and reference filgrastim (p = 0.001 and p = 0.020, respectively). No difference was seen between reference filgrastim and pegfilgrastim.
No differences concerning FN complications were seen (Table 3), with the exception of more chemotherapy delays in the biosimilar group when compared with pegfilgrastim (p = 0.040, post hoc analysis) but not with reference filgrastim.
Discussion
We aimed to add information, within the clinical practice setting, on the comparative effectiveness of biosimilar filgrastim (Nivestim™) in FN prevention in early breast cancer patients undergoing high-risk FN chemotherapy.
Our results showed a similar rate of FN in reference and biosimilar filgrastim groups (16 %). Even though the FN rate in the pegfilgrastim group was lower (9 %) than the reference filgrastim and also biosimilar groups, a substantial difference could not be demonstrated. This is similar to what was previously reported in head-to-head trials comparing reference filgrastim with pegfilgrastim [17, 18].
An interesting result of our study was a higher percentage of profound neutropenia (ANC < 100 cells/μL) at FN diagnosis in biosimilar filgrastim patients (50 vs. 4–6 % in the other two groups). The clinical relevance of this finding needs further confirmation, particularly in other G-CSF indications with increased risk of neutropenia like the bone marrow transplant setting where Nivestim™ obtained approval by extrapolation [19]. National Comprehensive Cancer Network (NCCN) guidelines mention profound neutropenia as a risk factor for developing infection-associated complications [11], yet other authors only consider profound neutropenia as a risk factor for FN worse outcome when combined with anticipated prolonged duration (>7 days) [20]. In our data, we could not accurately evaluate the duration of profound neutropenia; however, there were no differences between the three groups regarding FN hospitalization, septic shock or death. In fact, in the Nivestim™ pivotal trial [13], patients on the biosimilar arm also showed a greater proportion of severe neutropenia (defined as ANC <500 cells/μL) in the first two cycles. The clinical significance of this finding was an object of discussion by the EMA Committee for Medicinal Products for Human Use (CHMP) in the Nivestim™ pre-marketing assessment [19]. Again, as no detrimental effect in clinical parameters like FN incidence or number of infections was seen in biosimilar-treated patients, CHMP finally issued a favourable opinion.
The approval of Nivestim™ was supported by a single equivalence phase III trial [13]. Study allocation ratio was 2:1 and included 184 patients in the biosimilar filgrastim arm and 95 in the Amgen reference filgrastim group. Unlike our study that only included patients from the adjuvant/neoadjuvant context, this trial also included 16 % stage IV patients, which are considered a distinct population in terms of susceptibility for FN [21, 22]. Moreover, contrary to the pivotal trial where patients were only evaluated during the first three cycles of chemotherapy, our analysis comprised the entire duration of chemotherapy allowing us to assess the effectiveness of biosimilar filgrastim through a longer period.
Our FN rate in both reference filgrastim and pegfilgrastim cohorts is coincident with the one previously reported by the GEPARTRIO study [8] involving TAC regimen. On the other hand, our FN rate per biosimilar filgrastim was higher than the reported in the biosimilar pivotal trial [13] (16 % in our series vs. 2 %, both per patient). No major differences in filgrastim schedule and number of administrations existed between the two studies; thus, potential causes could include either a different FN definition or employment of a distinct anthracycline-taxane regimen. We fear that the FN incidence originally presented may underestimate the real FN rate in this population.
Weaknesses related to the retrospective nature of our study could be though pointed out, namely, the fact that patients in the biosimilar cohort were younger. Older age, especially above 60 years, is a risk factor for FN in breast cancer [23]. There were also differences concerning the use of ciprofloxacin in FN prophylaxis, which was higher in the biosimilar cohort in the first chemotherapy cycle. This seemed to have resulted in additional FN protection in the biosimilar group, since the cohort had lower FN rate in the first cycle (data not shown). Even though antibacterial prophylaxis in solid tumour conventional chemotherapy is not recommended [24], some data point towards a beneficial effect [25]. Altogether, these two imbalances could have acted at best as an advantage in the biosimilar cohort in what regards the FN rate.
Another possible bias was the difference in the proportion of patients treated in the neoadjuvant setting (approximately half of the patients in the pegfilgrastim and biosimilar cohorts as opposed to none in the reference filgrastim cohort). This indicates that a higher proportion of patients in pegfilgrastim and biosimilar group had larger breast cancer tumours at the time of treatment. Although evidence exists for considering advanced disease as a risk factor for FN [21, 22], it is unknown in what degree an advanced but only local and operable breast cancer could contribute to immunosuppression and act as an FN risk factor in an otherwise group of women with good performance status.
It should be noticed that no relevant discrepancies were, however, seen in important potential confounding factors like FN management protocols used over time, patterns of FN infectious pathogens or TAC dose intensity. Finally, our study did not address safety issues related with G-CSF use. This is because we felt that a retrospective chart analysis would never be fully comprehensive in respect of G-CSF toxicity evaluation. Therefore, we only focused on the G-CSF effectiveness data concerning FN episodes.
Biosimilars have their own complex manufacturing process which does not completely overlap the process of the reference medicine. Differences can be found and minor alterations in the various stages of the production may impact biosimilar activity and can go initially unnoticed due to limited clinical experience at approval. Post-approval data report becomes therefore essential. We did not detect differences in Nivestim™ effectiveness; however, further studies with larger number of patients and in other therapeutic settings are needed. Safety and also efficacy issues are currently being addressed by two ongoing Nivestim™ phase IV studies [26].
References
Kuderer NM, Dale DC, Crawford J et al (2006) Mortality, morbidity, and cost associated with febrile neutropenia in adult cancer patients. Cancer 106:2258–2266
Courtney DM, Aldeen AZ, Gorman SM et al (2007) Cancer associated neutropenic fever: clinical outcome and economic costs of emergency department care. Oncologist 12:1019–1026
Elting LS, Lu C, Escalante CP, Giordano SH et al (2008) Outcomes and cost of outpatient or inpatient management of 712 patients with febrile neutropenia. J Clin Oncol 26:606–611
Bonadonna G, Valagussa P (1981) Dose-response effect of adjuvant chemotherapy in breast cancer. N Engl J Med 304:10–15
Chirivella I, Bermejo B, Insa A et al (2009) Optimal delivery of anthracycline-based chemotherapy in the adjuvant setting improves outcome of breast cancer patients. Breast Cancer Res Treat 114:479–484
Martin M, Pienkowski T, Mackey J et al (2005) Adjuvant docetaxel for node-positive breast cancer. N Engl J Med 352:2302–2313
Martín M, Seguí MA, Antón A et al (2010) Adjuvant docetaxel for high-risk, node-negative breast cancer. N Engl J Med 363:2200–2210
Von Minckwitz G, Kümmel S, du Bois A et al (2008) Pegfilgrastim ± ciprofloxacin for primary prophylaxis with TAC (docetaxel/doxorubicin/cyclophosphamide) chemotherapy for breast cancer. Results from the GEPARTRIO study. Ann Oncol 19:292–298
Aapro MS, Bohlius J, Cameron DA et al (2011) 2010 update of EORTC guidelines for the use of granulocyte-colony stimulating factor to reduce the incidence of chemotherapy-induced febrile neutropenia in adult patients with lymphoproliferative disorders and solid tumours. Eur J Cancer 47:8–32
Crawford J, Caserta C, Roila F (2010) Hematopoietic growth factors: ESMO Clinical Practice Guidelines for the applications. Ann Oncol 21:248–251
Crawford J, Armitage J, Balducci L et al (2013) Myeloid growth factors. National Comprehensive Cancer Network. J Natl Compr Cancer Netw 11:1266–1290
Rajan SS, Carpenter WR, Stearns SC, Lyman GH (2013) Short-term costs associated with primary prophylactic G-CSF use during chemotherapy. Am J Manag Care 19:150–159
Waller CF, Semiglazov VF, Tjulandin S et al (2010) A phase III randomized equivalence study of biosimilar filgrastim versus Amgen filgrastim in patients receiving myelosuppressive chemotherapy for breast cancer. Onkologie 33:504–511
Passos-Coelho JL, Esteves S, Viera PA et al (2011) Adjuvant chemotherapy with TAC (docetaxel, doxorubicin, and cyclophosphamide) in patients with breast cancer–incidence of neutropenic fever outside clinical trials. Breast J 17:539–541
Hryniuk WM (1988) The importance of dose intensity in the outcome of chemotherapy. In: DeVita VT, Hellman S, Rosenburg SA (eds) Important advances in oncology. JB Lippincott, Philadelphia, pp 121–141
Product Monograph Neupogen: https://www.amgen.ca/Neupogen_PM.pdf
Holmes FA, Jones SE, O’Shaughnessy J et al (2002) Comparable efficacy and safety profiles of once-per-cycle pegfilgrastim and daily injection filgrastim in chemotherapy-induced neutropenia: a multicenter dose-finding study in women with breast cancer. Ann Oncol 13:903–909
Green MD, Koelbl H, Baselga J et al (2003) A randomized double-blind multicenter phase III study of fixed-dose single-administration pegfilgrastim versus daily filgrastim in patients receiving myelosuppressive chemotherapy. Ann Oncol 14:29–35
EMA Nivestim™ assessment report: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/001142/WC500093664.pdf
Freifeld AG, Bow EJ, Sepkowitz KA et al (2011) Infectious Diseases Society of America. Clinical practice guideline for the use of antimicrobial agents in neutropenic patients with cancer: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis 52(4):427–431
González-Barca E, Fernández-Sevilla A, Carratalá J et al (1999) Prognostic factors influencing mortality in cancer patients with neutropenia and bacteremia. Eur J Clin Microbiol Infect Dis 18:539–544
Hosmer W, Malin J, Wong M (2011) Development and validation of a prediction model for the risk of developing febrile neutropenia in the first cycle of chemotherapy among elderly patients with breast, lung, colorectal, and prostate cancer. Support Care Cancer 19:333–341
Dranitsaris G, Rayson D, Vincent M et al (2008) Identifying patients at high risk for neutropenic complications during chemotherapy for metastatic breast cancer with doxorubicin or pegylated liposomal doxorubicin: the development of a prediction model. Am J Clin Oncol 31:369–374
Flowers CR, Seidenfeld J, Bow EJ et al (2013) Antimicrobial prophylaxis and outpatient management of fever and neutropenia in adults treated for malignancy: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 31:794–810
Timmer-Bonte JN, de Boo TM, Smit HJ et al (2005) Prevention of chemotherapy-induced febrile neutropenia by prophylactic antibiotics plus or minus granulocyte colony-stimulating factor in small-cell lung cancer: a Dutch randomized phase III study. J Clin Oncol 23:7974–7984
Kamioner D, Fruehauf S, Maloisel F et al (2013) Study design: two long-term observational studies of the biosimilar filgrastim Nivestim™ (Hospira filgrastim) in the treatment and prevention of chemotherapy-induced neutropenia. BMC Cancer 13:547
Acknowledgments
The study was performed while the third author was employed at the original institution (first affiliation), but this author has recently moved to a new employer (third affiliation).
Funding
None.
Conflict of interest
The authors have declared no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Brito, M., Esteves, S., André, R. et al. Comparison of effectiveness of biosimilar filgrastim (Nivestim™), reference Amgen filgrastim and pegfilgrastim in febrile neutropenia primary prevention in breast cancer patients treated with neo(adjuvant) TAC: a non-interventional cohort study. Support Care Cancer 24, 597–603 (2016). https://doi.org/10.1007/s00520-015-2818-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00520-015-2818-2