
Abstract
A class of ribonucleases termed S-RNases, which control the pistil expression of self-incompatibility, represents the only known functional products encoded by the S locus in species from the Solanaceae, Scrophulariaceae and Rosaceae. Previously, we identified a pollen-specific F-box gene, AhSLF (S locus F-box)-S 2, very similar to S 2 -RNase in Antirrhinum, a member of the Scrophulariaceae. In addition, AhSLF-S 2 also detected the presence of its homologous DNA fragments. To identify these fragments, we constructed two genomic DNA libraries from Antirrhinum self-incompatible lines carrying alleles S 1 S 5 and S 2 S 4, respectively, using a transformation-competent artificial chromosome (TAC) vector. With AhSLF-S 2-specific primers, TAC clones containing both AhSLF-S 2 and its homologs were subsequently identified (S 2 TAC, S 5 TACa, S 4 TAC, and S 1 TACa). DNA blot hybridization, sequencing and segregation analyses revealed that they are organized as single allelic copies (AhSLF-S 2, -S 1, -S 4 and -S 5) tightly linked to the S-RNases. Furthermore, clusters of F-box genes similar to AhSLF-S 2 were identified. In total, three F-box genes (AhSLF-S 2, -S 2 A and -S 2 C) in S 2 TAC (51 kb), three (AhSLF-S 4, -S 4 A and -S 4 D) in S 4 TAC (75 kb), two (AhSLF-S 5 and -S 5 A) in S 5 TACa (55 kb), and two (AhSLF-S 1 and -S 1 E) in S 1 TACa (71 kb), respectively, were identified. Paralogous copies of these genes show 38–54% identity, with allelic copies sharing 90% amino acid identity. Among these genes, three (AhSLF-S 2 C, -S 4 D and -S 1 E) were specifically expressed in pollen, similar to AhSLF-S 2, implying that they likely play important roles in pollen, whereas three AhSLF-SA alleles showed no detectable expression. In addition, several types of retroelements and transposons were identified in the sequenced regions, revealing some detailed information on the structural diversity of the S locus region. Taken together, these results indicate that both single allelic and tandemly duplicated genes are associated with the S locus in Antirrhinum. The implications of these findings in evolution and possible roles of allelic AhSLF-S genes in the self-incompatible reaction are discussed in species like Antirrhinum.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In flowering plants, a widely used mechanism to prevent inbreeding is known as self-incompatibility (SI), which is, in most cases, controlled by a single multi-allelic locus, the S locus (de Nettancourt 2001). Recent biochemical and molecular studies have identified genes encoded by the S locus in several plant families. In the Brassicaceae, two genes known as SRK expressed in the stigma and SCR/SP11 expressed in pollen (Stein et al. 1991; Schopfer et al. 1999; Suzuki et al. 1999) belong to one haplotype and have been shown to control pollen recognition through a ligand-receptor binding mechanism (Kachroo et al. 2001; Takayama et al. 2001). In the Papaveraceae, a pistil-specific gene (S 1) is encoded by the S locus (Foote et al. 1994), and a sophisticated mechanism involving Ca2+ signaling is involved in pollen tube growth inhibition (Wheeler et al. 2001). In self-incompatible species from the Solanaceae, Scrupholariaceae and Rosaceae, a group of ribonucleases termed S-RNases are encoded by their respective S loci and control self-pollen recognition and rejection by the pistil (Anderson et al. 1986; McClure et al. 1989; Ai et al. 1990; Broothaerts et al. 1995; Sassa et al. 1996; Xue et al. 1996; McCubbin and Kao 2000).
However, because of the elusive identity of pollen S gene (Sp), it is not clear how S-RNase functions during the self-incompatible reaction. Nevertheless, several recent studies have indicated that S-RNases likely interact with an inhibitor inside the pollen tube to accomplish pollen recognition and growth inhibition (Golz et al. 1999, 2001; Luu et al. 2001). In Nicotiana and Petunia, S allele duplications have been found to be associated with pollen-part self-compatible mutants (Pandy 1965; Golz et al. 1999). The genetic behavior of these mutants can be explained if Sp acts as an inhibitor of all S-RNases except its cognate S-RNase (Golz et al. 2000). This model is further supported by the finding that S-RNase uptake by the pollen tube has no allelic specificity (Luu et al. 2000). Recently, Luu et al. (2001) have found that pollen containing two different S alleles is accepted by S 11/S 13 style in Solanum chacoense, but is rejected by a chimeric S11/13 RNase, indicating that Sp genes must be expressed in diploid heteroallelic pollen, and proposed that Sp consists of two components, a general S-RNase inhibitor and an S-allele-specific product blocking the inhibitory action on self-RNases. Several groups have attempted to identify the Sp genes in Nicotiana alata, Petunia inflata, Antirrhinum and two Prunus species (Li et al. 2000; McCubbin et al. 2000; Lai et al. 2002; Entani et al. 2003; Ushijima et al. 2003). Although none of these candidate Sp have been demonstrated to play a role in SI, interestingly, Lai et al. (2002) identified a pollen-specific F-box gene, AhSLF-S 2, located 9 kb away from S 2 -RNase, indicating a possible involvement of a protein degradation pathway in the self-incompatible reaction in species like Antirrhinum. In the same study, genomic DNA fragments homologous to AhSLF-S 2 were also detected but their relationships were not clear (Lai et al. 2002). Recently, several pollen-expressed F-box genes similar to AhSLF-S 2 have also been found in the S locus of both almond (Prunus dulcis) and Japanese apricot (Prunus mume) with a haplotype-specific polymorphism, indicating that they are good Sp candidates (Entani et al. 2003; Ushijima et al. 2003).
As shown by their presence in a wide range of flowering plants and phylogenetic analyses of S-RNases, the S loci of the Solanaceae, Scrophulariaceae and Rosaceae likely share a common ancestor and probably predate eudicot diversification (Igic and Kohn 2001; Steinbachs and Holsinger 2002). This ancient origin may eventually have resulted in a complex genomic structure of the S locus. In fact, initial genomic structural analysis of the S locus revealed that it is located in a region consisting of repetitive sequences (Coleman and Kao 1992; Royo et al. 1996; Lai et al. 2002; Entani et al. 2003; Ushijima et al. 2003). As yet, limited information is available on the genomic constituents of the S locus in S-RNase-based self-incompatible species.
To study the genomic structure of the S locus and the relationship between AhSLF-S 2 and its homologues, we have constructed two genomic DNA libraries of Antirrhinum with different alleles using a transformation-competent artificial chromosome (TAC) vector. We identified TAC clones containing AhSLF-S 2 and its homologues and subsequently demonstrated that they are single allelic genes. DNA sequence analysis revealed the presence of several different types of retroelements and transposons near S-RNase genes. Furthermore, clusters of F-box genes homologous to AhSLF-S 2 were detected and their expression and genomic organization analyzed. The implications of the similarity between SLF genes and some F-box genes in Arabidopsis and tomato and the possible functions for AhSLF-S gene in the SI reaction in species like Antirrhinum are discussed.
Materials and methods
Plant materials
Self-incompatible lines derived from an interspecific cross between Antirrhinum hispanicum and Antirrhinum majus, as well as their growth conditions were described previously (Xue et al. 1996). Two SI lines with S 1 S 5 and S 2 S 4 alleles were further crossed to generate a progeny population of 100 plants segregating for four S alleles.
Construction and screening of TAC library
High-molecular-weight (HMW) DNA of over 2 Mb from two SI lines with S 2 S 4 and S 1 S 5 alleles was prepared from leaf nuclei according to Liu and Whitter (1994). Partial digestion of HMW-DNA with HindIII, TAC vector preparation, ligation and transformation of Escherichia coli DH10B by electroporation followed the method described by Liu et al. (2000). The HindIII-digested TAC plasmid DNA was sized-fractioned with a field inversion agarose gel electrophoresis to check the insert length. A total of ca. 77,925 recombinant clones were selected and stored in 384-well plates.
For each library, the clones of one 384-well plate were imprinted onto a 15 cm plate with a VP384 pin (V&P Scientific, San Diego, Calif.) and inoculated onto LB agar medium containing kanamycin (25 mg/l). After incubation at 37°C overnight, the bacteria on the 15 cm plate were collected for plasmid preparation. Plasmid DNA from ten 384-well plates was mixed as a pool for PCR screening. The TAC library was screened with the primers specific for S 2, S 4 and S 5 -RNase (Xue et al. 1996), and G11E and G11D (Lai et al. 2002). Once a specific PCR product was detected in one or more pools, the ten 384-well plates of the positive pool were individually screened with the primer pair again. A positive 384-well plate was subsequently identified. Finally, the positive clone was identified by PCR screening in a row and column combination. S 2- (G2338 and G1222), S 4- (G3169 and G1224), and S 5 -RNase primers (G2339 and G1481) were according to Xue et al. (1996). AhSLF-S 2 primers (G11E, G11D, G11f and G11j) were described by Lai et al. (2002). The 3′ UTR region-specific primers were as follows: AhSLF-S 5 (CGGAGTGTCGGTGCATCATAG), AhSLF-S 4 (ACTTAACCAACTCGGATTGAA) and AhSLF-S 1 (TCATAATTTAAACCCGCCACC).
Sequencing and assembling of TAC clones
TAC clone sequencing and assembly was carried out as previously described by Lai et al. (2002).
Southern and northern blotting analyses
Genomic DNA isolation was performed as previously described (Xue et al. 1996). DNA (10 μg) was digested, separated on a 0.8% agarose gel and transferred onto Hybond N+ (Amersham, Piscataway, N.J.) membrane. Prehybridization, hybridization and washing of the blot were performed as recommended by the manufacturer. Total RNA was extracted from different tissues using an RNeasy Plant Mini Kit (Qiagen, Hilden, Germany). RNA samples were separated on 1% agarose/formaldehyde gels and transferred to Hybond N+. Prehybridization, hybridization and washing of the blot were performed as recommended by the manufacturer. Probes were labeled with 32P by random priming using the Prime-a-Gene labeling system (Promega, Madison, Wis.).
Reverse transcription-PCR analysis
Total RNA was prepared as previously described and was digested with DNase I (Takara, Kyoto, Japan). Reverse transcriptase (RT) (Invitrogen, Carlsbad, Calif.) was used to synthesize first strand cDNA. RT-PCR primers were designed from the full-length coding sequences of F-box genes.
Sequence annotation and computational analysis
Genescan and FgeneSH softwares were used for gene prediction (http://www.ncgr.ac.cn). BLASTx, BLASTp and BLAST2 (http://www.ncbi.ac.cnI) and WU-BLAST2 and CLUSTALW (http://www.ebi.ac.uk) were used for DNA sequence analysis. A Dotter program (http://www.cgr.ki.se) was used for comparative analysis of TAC insert sequences. The phylogenetic tree was generated with CLUSTALW using a neighbor-joining feature (http://www.ebi.ac.uk/).
Results
Isolation of genes homologous to AhSLF-S 2
An F-box gene, AhSLF-S 2, about 9 kb away from the S 2 -RNase gene was previously identified and its homologues detected in Antirrhinum (Lai et al. 2002). However, it was not clear whether these represent allelic or duplicated copies of the same gene. To resolve these possibilities, we constructed two genomic DNA libraries from self-incompatible lines of S 2 S 4 and S 1 S 5 genotypes using the vector TAC7 (Liu et al. 1999). The two libraries had 39,936 and 38,016 clones, respectively; both had an estimated average insert length of 70 kb (data not shown), and were equivalent to 6 and 5.8 times the haploid genome of Antirrhinum. Initially, we suspected that AhSLF-S 2 homologues should be tightly linked to S-RNase genes as had been found for AhSLF-S 2. Thus, we isolated TAC clones containing S-RNases using a pooled PCR method in which plasmid DNA was individually prepared from each 384-well plate and the plasmid DNA from ten 384-well plates was pooled together for PCR screening with gene-specific primers. In total, we identified one TAC clone containing S 2 -RNase (S 2 RNaseTAC), four containing S 4 -RNase (S 4 RNaseTACa–d) and two containing S 5 -RNase (S 5 RNaseTACa, b) (Fig. 1). DNA blot hybridization and sequencing analysis of S 4 -RNaseTACa and S 5 -RNaseTACa revealed that they did not contain AhSLF-S 2 homologues, indicating that the latter are not as close to S-RNases as is the case in the S 2 haplotype (J. Zhou, F. Wang and Y. Xue, MS in preparation).

Transformation-competent artificial chromosome (TAC) physical maps corresponding to four S alleles. The sequenced region corresponding to S 2 allele represents 110 kb in length, and S 2-RNase and AhSLF-S 2 are separated by ca. 9 kb. In the TAC contigs corresponding to the three other alleles, the physical distance between S-RNase and S TAC clones is not known. TAC clones corresponding to the S-locus F-box (SLF) regions are aligned based on homology. Expressed or predicted AhSLF genes are indicated as solid bars. The probes used in DNA blot analysis are shown as black bars. * Completely sequenced TAC clones
To further identify AhSLF-S 2 homologues, we postulated that they should be highly similar in sequences, based on the cross DNA hybridization (Lai et al. 2002), therefore two primers (G11E and G11D) were designed based on the coding region and used to amplify the TAC libraries with the pooled PCR method. In total, five clones were obtained from the S 1 S 5 library and two clones from the S 2 S 4 library. To classify them, 1 kb PCR products amplified by the primers from each clone were sequenced. The results showed they share over 97% identity and can be organized into four different groups, with two groups from each SI line. In the five clones from the S 1 S 5 library, two distinct sequences were found, represented by three (S 5 TACa–c) and two (S 1 TACa, b) TAC clones with identical sequence, respectively (see Fig. 1). The two clones from the S 2 S 4 library (S 2 TAC and S 4 TAC) were different from each other, but the sequence of S 2 TAC was identical to that of AhSLF-S 2 (see Fig. 1). These results suggest that AhSLF-S 2 homologous sequences from the four groups of the TAC clones are alleles representing four S haplotypes (also see below).
Gene content and structure of the SLF regions
To investigate the structure of the SLF regions, four TAC clones (S 2 TAC, S 5 TACa, S 4 TAC and S 1 TACa) were selected and fully sequenced. Their insert lengths were 51, 55, 75 and 71 kb, respectively (see Fig. 3). The total sequenced region combining S 2 BAC (Lai et al. 2002) and S 2 TAC is 110 kb in length. Sequence analysis of S 2 TAC revealed that it has a copy of AhSLF-S 2. Based on linkage analysis, S 4 TAC, S 1 TACa and S 5 TACa represent S 4, S 1 and S 5 alleles, respectively (see Fig. 4). However, S 4 TAC and S 5 TACa did not contain S 4- and S 5-RNase genes (see Fig. 1), confirming that the distances between AhSLF-S 2 homologues and S-RNases in these two alleles are much larger than those in S 2.
To examine gene content in the four TAC sequences, gene prediction analysis revealed that, on average, two-to-three predicted genes were found to be homologous to AhSLF-S 2 in each sequenced region, in addition to transposable elements (see Fig. 1 and Table 1). In S 2 TAC, two additional SLF genes (SLF-S 2 A and S 2 C) were predicted. Three SLF genes were detected in S 4 TAC, two in S 5 TACa and two in S 1 TACa. To reveal the identity among AhSLF-S genes, we aligned the predicted polypeptide sequences of the four AhSLF-S genes (Fig. 2A). The results showed that AhSLF-S 2 and its homologues, termed AhSLF-S 1, S 4 and S 5, share more than 97% identity at the amino acid level, indicating that they are allelic (Fig. 2A). AhSLF-S 2 A and its homologues share more than 90% identity and appear to be allelic (data not shown), and were named AhSLF-S 4 A and S 5 A. However, the other three predicted genes in this region have only 42–48% identity either between themselves or with AhSLF-S, indicating that they are not allelic copies. These were named AhSLF-S 2 C, -S 4 D and -S 1 E (Fig. 2B, Table 2). In addition, within the same haplotype, predicted SLF proteins have about 38–54% identity, indicating that their duplications were ancient. Together, these results showed that paralogous SLF genes in each allele are organized as clusters, and that AhSLF-S 2 and -S 2 A and their homologues appear to be allelic.


Amino acid sequence alignment of predicted SLF polypeptides. A Deduced sequences of AhSLF-S alleles were compared. B Sequences were derived from Antirrhinum (AhSLF-S 2 , -S 2 C, -S 4 D and -S 1 E), Prunus dulcis (PdSFBa, PdSFBb and PdSLFc), Prunus mume (PmSLF-S 1 , PmSLF-S 7 and PmSLFL 1 -S 7 ) and Arabidopsis (At4g12560 and At4g22390)
To examine the relationship of the known SLF genes in the S locus, polypeptides from Antirrhinum and two roceaous species were aligned. The similarity between SLF genes in Antirrhinum and Rosaceae is very low, ranging from 15 to 25%, indicating they have been separated for an extremely long time (Fig. 2B, Table 2). Nevertheless, the conserved amino acids in their F-box domains suggest that they all belong to the same F-box family. In addition, all of them have some conserved regions besides the F-box domain that are also evident in similar predicted polypeptides from Arabidopsis (Fig. 2B).
Many retroelements or transposons were identified in the sequenced genomic regions covered by these TAC clones (Table 1). In total, 5, 2, 5 and 4 predicted genes of S 1-, S 2-, S 4- and S 5 -TAC, respectively, represented retroelements or transposons. The remainder of the predicted genes in these sequenced regions have no identity to any known genes in the EMBL database.
To classify the predicted retroelements and transposons, BLASTx was used to compare these sequences in detail. In total, six non-LTR LINE-like elements were identified, named L 1 –L 6 (Table 3, Fig. 3). These elements are characterized by the presence of a polyprotein, a 3′ poly (A) signal, and a 5 bp target site insertion signature (Table 3). Additionally, nine predicted retroelements (R 1 –R 9) showed no typical LTRs or non-LTR features (Fig. 3), and likely represent aberrant or scrambled retrotransposons. Finally, two putative transposon proteins were predicted in S 4 TAC, with 57% amino acid identity to TNP2-like (Nacken et al. 1991), and 56% identity to En/Spm-like transposon (Frey et al. 1989), respectively (Table 1). These results showed that the SLF regions are enriched with both non-LTR retrotransposons, consistent with its location near the centromere (Ma et al. 2002) and a feature shared with other RNase-based self-incompatible species (Ushijima et al. 1998; Entani et al. 1999).

Genomic structural comparisons of the SLF regions in Antirrhinum. The S 2 allele region was derived by combining both S 2 BAC (Lai et al. 2002) and S 2 TAC sequences. Non-LTR LINE elements, putative retroelements and En/Spm-like transposon protein are indicated with lines of different colors. The genes are shown with red, purple or green lines; arrows transcriptional direction. Regions of duplication/deletions occurring between the S 2 allele and S 1, S 4 or S 5 alleles are indicated by solid lines, and those between S 1, S 4 and S 5 alleles are indicated by dashed lines
To identify the structural relationship between the SLF regions, we used a Dotter program to conduct a detailed pairwise comparative analysis. Despite the overall nucleotide similarity over the most regions, several major duplications and insertion/deletions (indels) were identified (Fig. 3). Comparing S 2 TAC with S 1 TACa, S 4 TAC or S 5 TACa, a notable difference among the four regions sequenced was related to a 2 kb sequence near the left end of S 2 TAC, which was deleted in all the other three TAC clones, suggesting that insertions have occurred between S-RNase and AhSLF-S genes in these three regions. Another variable region was a 5 kb sequence of S 2 TAC that was deleted in S 5 TACa, and also a 2 kb sequence of S 2 TAC (29–31 kb) was repeated in S 4 TAC. Several differences were also identified when comparing S 1 TACa, S 4 TAC and S 5 TACa. Reciprocal deletions of 5 kb (42–47 kb) and 8 kb sequence (52–60 kb) of S 5 TAC were detected between S 1 TACa and S 5 TACa (Fig. 3). As a result, retroelements R 1 and R 8 were predicted in S 1 TACa and S 5 TACa, respectively, but R 8 of S 5 TACa was 6 kb smaller than R 1 of S 1 TACa. Also, an extra retroelement (R 7) was detected in S 5 TACa but not in S 1 TACa due to deletion of a 12 kb sequence in S 1 TACa. A second variable region corresponded to an 8 kb sequence of S 4 TAC, which was deleted in S 1 TACa and S 5 TACa or vice versa. Subsequently, the two putative transposons were predicted only in S 4 TAC. A third apparent difference was detected between S 4 TAC and S 5 TACa. A 10 kb region of S 4 TAC was deleted in S 5 TACa. Two non-LTR LINE-like elements (L 1 and L 2) were predicted in S 1 TACa only, and in this region S 1 TACa has no overlap with other TAC clones, indicating a breakdown of synteny. Interestingly, this region is located to the right of the non-allelic AhSLF genes, implying that the latter likely resulted from unequal crossovers. Taken together, in addition to the occurrence of retrotransposition events, indels and duplications are also associated with the S locus region, indicating that these events have played important roles in its evolution.
The close linkage of SLF genes to the S locus
Although the genomic structures of the SLF regions showed some variations among S haplotypes, the linkage of AhSLF-S 2 to S 2 -RNase indicated that the other AhSLF-S alleles are also probably linked to the S locus. To determine their relationship with the S locus, the sequences of the four AhSLF-S genes were aligned (data not shown). The coding regions were highly homologous, but differences were observed in the 3′-UTR regions. Thus, we designed a common upstream primer (G11j) from the conserved coding region and individual downstream primers generated from the 3′-UTR regions of each AhSLF-S gene. Subsequently, the four pairs of gene-specific primers were used to analyze the linkage between S-RNase and AhSLF-S genes.
We generated a population of over 100 progeny segregating for four S alleles by crossing two self-incompatible S 1 S 5 and S 2 S 4 lines. Genomic DNA of 100 progeny and parental plants was used for PCR analysis with specific primers for S 2-, S 4- and S 5 -RNase and the four pairs of primers for AhSLF-S genes. Representative PCR results are shown in Fig. 4. S 2 , S 4 and S 5 genotypes were determined using S-RNase-specific primers and S 1 was inferred by absence of the PCR products of the other three S-RNases (Fig. 4). The PCR products representing S 5-, S 4- and S 2-RNases were about 1 kb, 2 kb and 1 kb, respectively (Fig. 4). By using AhSLF-S gene-specific primers, we detected an absolute correlation between AhSLF-S alleles and their respective S-RNases, showing that they are tightly linked to each other. The results of χ 2 test (n=3,χ 2=1.55, P<0.05) showed that the four genes were segregated as 1:1:1:1 and perfectly correlated to S-RNase gene segregation, implying that they transmit as a single locus. In all the plants inferred to have S 1 genotype, a specific product of 0.4 kb was detected using AhSLF-S 1 primers. These results clearly show that AhSLF-S genes are tightly linked to the S locus, and possibly inherited as a haplotype together with S-RNase genes. Therefore, we performed a phylogenetic analysis based on the deduced amino acid sequences of the AhSLF-S genes. The topology of the phylogeny of AhSLF-S agreed with that of three Antirrhinum S-RNases (data not shown), suggesting that these two genes are under similar selection pressure.

AhSLF-S genes are tightly linked to their respective S-RNase genes. PCR was performed on genomic DNA from an S allele segregating population with gene-specific primers. The sizes of amplified products are indicated on the right. Two lanes on the left represent the parental plants. The ten lanes on the right represent progeny plants. The S genotypes are indicated on the top of the panel. A–D PCR amplification with specific primers for S 2-RNase and AhSLF-S 2, S 4-RNase and AhSLF-S 4, S 5-RNase and AhSLF-S 5 and AhSLF-S 1, respectively
SLF genes are specifically expressed in pollen
Because the newly identified SLF genes were predicted, it was not clear whether they are expressed. To investigate their expression, primers derived from AhSLF-S alleles AhSLF-S 2 A, -S 2 C, -S 4 D and -S 1 E were used for RACE analysis. The results showed that these AhSLF-S alleles are specifically expressed in pollen containing either S 1 S 5 or S 2 S 4 alleles (data not shown), similar to AhSLF-S 2 (Lai et al. 2002). RACE products matching the genomic sequences of AhSLF-S 2 C, -S 4 D and -S 1 E were obtained and sequenced, and the predicted proteins have 384, 374 and 384 amino acids, respectively (see Fig. 2B). However, no RACE products were detected for AhSLF-S 2 A in any tissue (data no shown), indicating either that it is expressed to levels below the current detection limit or that it is not expressed under the conditions tested.
To further confirm the expression of these genes, RT-PCR analysis was conducted using primers derived from their full-length coding regions. The templates consisted of genomic DNA, or cDNA synthesized with or without RT (reverse transcriptase) from RNA extracted from leaf, stigma, petal and pollen (with S 1 S 5 or S 2 S 4 alleles). As shown in Fig. 5, PCR products of ca. 1.1 kb were exclusively detected in pollen derived from both S 1 S 5 and S 2 S 4 alleles, consistent with the presence of similar allelic transcripts. DNA sequencing of the 1.1 kb PCR products further confirmed that they were derived from AhSLF-S 2 C, S 4 D and S 1 E or their alleles (data not shown, but see Fig. 6). Similarly sized fragments were detected in genomic DNA, showing that these genes are intronless, consistent with the predicted gene models. These results indicate that Antirrhinum pollen expresses several SLF genes with a pattern similar to that of AhSLF-S 2 (Lai et al. 2002). In addition, the absence of AhSLF-S 2 A expression in pollen rules out the possibility that it encodes Sp.

Expression of AhSLF-S 2 C, -S 4 D and -S 1 E. cDNA were synthesized with (+) or without (−) reverse transcriptase (RT) from total RNA from leaf, stigma, petal, and pollen (S 1 S 5 and S 2 S 4 alleles). As a control, PCR was also performed on genomic DNA (gDNA). The full-length coding regions of about 1.1 kb were detected with the specific primers for each of the three genes. Tubulin cDNA was amplified as a control

Genomic organization of AhSLF genes. Genomic DNA from various S alleles containing lines was digested with HindIII, EcoRI or BamHI and separated by agarose gel electrophoresis. The genotypes of the plants are indicated on the top. The numbers indicate the sizes of hybridizing fragments in kb. Probes used in A–D were the predicted coding sequences derived AhSLF-S 2 A, -S 2 C, -S 1 E and -S 4 D, respectively
Genomic organization of the SLF genes
To reveal the genomic organization of the SLF genes, we performed genomic DNA blot analysis using the predicted coding region of AhSLF-S 2 A, S 2 C, S 4 D and S 1 E as probes (Fig. 6). Hybridization to AhSLF-S 2 A detected a 2.5 kb HindIII fragment in S 2-containing lines, as predicted from the S 2 TAC sequence. In addition, HindIII fragments of 9 kb and 2.3 kb were detected in S 4-containing, and in S 5- and S1-containing lines, respectively; also consistent with the predicted HindIII fragments from their respective TAC sequences (Fig. 6A). However, EcoRI digestion revealed no RFLP except in S 5-containing lines. These results demonstrated that AhSLF-S 2 A alleles are present in all the S-haplotypes as single copy genes.
AhSLF-S 2 C hybridized to an 8 kb HindIII fragment in S 2-containing lines, representing the S 2 allele (Fig. 6B). A specific 18 kb BamHI fragment was detected in S 2- and S 1-containing lines. A 12 kb BamHI fragment was detected in both S 1 S 5 and S 2 S 4 lines, likely representing both the S 5 and S 4 alleles. Based on the fact that there is neither a HindIII nor a BamHI recognition site in the coding region of AhSLF-S 2 C, the results indicate that AhSLF-S 2 C is a single copy gene that also has allelic copies.
AhSLF-S 1 E displayed no polymorphism in HindIII-digested DNA (Fig. 6C). However, a 3 kb EcoRI fragment was detected in S 1-containing lines as predicted from S 1 TACa sequence. In addition, another 4 kb EcoRI fragment was detected in all the S 2-containing lines. Therefore, these two fragments likely represent S 1 and S 2 alleles, respectively. Other EcoRI or HindIII fragments showed no clear polymorphism between lines but are likely derived from S 4 and S 5 alleles. It appeared that AhSLF-S 1 E is also a single copy gene.
Hybridization of HindIII-digested genomic DNA to AhSLF-S 4 D detected no allele-specific fragments except in S 5-containing lines (Fig. 6D). In EcoRI-digested DNA, a fragment of 14 kb was detected in lines containing S 4, as predicted from the S 4 TAC sequence. Another 10 kb EcoRI fragment was detected in the S 5-containing lines and a 1.2 kb fragment in S 1-containing lines, likely representing S 5 and S 1 alleles, respectively. The 2.5 kb fragments showed no polymorphism in these lines. In addition, the 1.8 and 1.6 kb EcoRI fragments showed no linkage to any haplotype. This result suggested that AhSLF-S 4 D is present in more than one copy, suggesting that it has been duplicated in the genome.
Taken together, the genomic organization of AhSLF-S 2 A, -S 2 C and -S 1 E suggested that they are all single copy genes. In contrast, AhSLF-S 4 D is divergent and there is more than one copy in the genome.
F-box genes similar to SLF are found in Arabidopsis and tomato
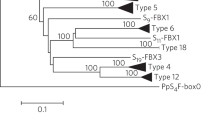
To examine the relationships of SLF genes, a phylogenetic analysis was performed based on deduced amino acid sequences from several available F-box genes or ESTs from Arabidopsis and tomato together with SLFs from Antirrhinum and Rosaceae (Fig. 7). The result showed that SLF genes are more closely related to each other, including an EST sequence from tomato (BI933580) expressed specifically in flowers. Although the functions of all of these F-box genes are still unknown, the results indicated that SLF genes belong to a very large F-box gene family with a wide distribution.

A phylogenetic tree of predicted SLF polypeptides. The sequences were from Antirrhinum (AhSLF-S2, AhSLF-S2C, AhSLF-S4D and AhSLF-S1E), Prunus dulcis (PdSFBa, PdSFBb and PdSLFc), Prunus mume (PmSLF-S1, PmSLF-S7 and PmSLFL1-S7), tomato (AW738697, BE462774, BI928765, BI933580, BI933949 and TC103260) and Arabidopsis (At2g43260, At3g06240, At3g07870, At3g23880, At4g12560 and At4g22390)
Discussion
By detailed molecular studies, we have identified four clusters of F-box genes tightly linked to the S locus in Antirrhinum. Importantly, AhSLF-S 2 and its homologs are present as single allelic genes with pollen-specific expression, supporting the possibility that they play a role in SI. In addition, gene duplications and the associations of non-LTR LINE-like elements and indels with the S locus region have provided some insights into its evolution.
Both allelic and tandemly repeated genes are associated with the S locus
It is well known that the S locus possesses a large number of alleles (de Nettancourt 2001). For example, over 37 alleles have been genetically identified in Oenothera organensis (Emerson 1939), 32 alleles in Papaver rhoeas (Lawrence et al. 1993), and over 40 alleles in Physalis crassifolia of the Solanaceae (Richman et al. 1996). Nevertheless, relatively little is known about how they were actually generated over time. The disease resistance R locus is another well-studied recognition locus in plants (Martin et al. 1993; Hammond-Kosack et al. 1998; Ellis et al. 2000a). R-locus-encoded genes have been found to occur either as simple (single allelic series) or, more often, complex loci consisting of duplicated genes (Ellis et al. 2000b). In both cases, similar evolutionary processes, including unequal crossing-over, gene conversion and diversifying selection, appear to contribute to overcoming rapid pathogen variations (Michelmore and Meyers 1998; Ellis et al. 2000a). The Antirrhinum S locus region appears to contain both types of gene organization, indicating that the evolutionary forces displayed by the R locus also are operating. So far, over 200 S-RNases, organized as allelic genes, have been identified in a range of different species (de Nettancourt 2001). In addition, our results have shown that clusters of paralogous AhSLF genes are closely associated with the S-RNases, a feature also shared by the rosaceous SLF genes (Entani et al. 2003; Ushijima et al. 2003). However, it is not clear whether these similarities between the S and R loci are intrinsic to their specific roles in terms of recognition. The mate recognition locus in yeast and the major histocompatibility complex (MHC) locus in animals also have similar features (May and Matzke 1995; O'hUigin 1995), supporting the view that they are intrinsic properties of recognition loci.
Structural diversity of the S locus region
The genomic region containing the S locus appears to be extremely diverse. Previous studies in several RNase-based self-incompatible species have shown that their S loci contain repetitive sequences (Coleman and Kao 1992; Royo et al. 1996; Lai et al. 2002; Entani et al. 2003; Ushijima et al. 2003), providing a structural basis for recombination suppression. Recently, Ma et al (2002) also found that the S locus of Antirrhinum is located in the pericentromeric region. Molecular studies have revealed that this region consists of abundant retroelements in several plant species, including Arabidospsis and rice (Copenhaver et al. 1999; Cheng et al. 2002). Consistent with this, we have found several types of retroelements, such as LINE-like protein elements and putative retrotransposons, as well as putative transposons associated with S-RNases in the S locus region in Antirrhinum (Fig. 3). In fact, these features have also been found to be associated with the R locus (Wei et al. 2002). For example, two nested complexes of transposable elements and a 45 kb tandem repeat region have been described in the Mla resistance locus in barley. The retroelements in this region belong to LTR retrotransposons and non-LTR LINE-like elements, which are thought to have played a role in recombination suppression over time (Duret et al. 2000; Fu et al. 2002; Rizzon et al. 2002).
Although it is difficult to estimate the actual time at which transposition events occurred in the S locus region due to the limited information available, what appears to be certain is that transposable elements and indels have played important roles in generating the structural diversity of the S locus region. Our data revealed that both the duplicated gene organization and dynamic genomic structures have contributed to the birth and persistence of allelic diversity of the S locus.
Are AhSLF genes capable of encoding Sp products?
An important unresolved issue in S-RNase-based SI is how S-RNases function inside the pollen tube. Two models, the gatekeeper model and the inhibitor model, have been proposed to explain this (Wheeler et al. 2001). Based on the evidence currently available, the inhibitor model is more favored. Although several Sp candidates have been isolated (Li et al. 2000; McCubbin et al. 2000; Lai et al. 2002; Entani et al. 2003; Ushijima et al. 2003), none have yet been assigned a role, if any, in SI. Nevertheless, the pollen-expressed SLF genes with haplotype-specific polymorphisms in P. dulis and P. mume represent good candidates for Sp (Entani et al. 2003; Ushijima et al. 2003).
Interestingly, we have identified a cluster of three F-box genes near the S 2 -RNase gene within a 70 kb region in Antirrhinum; two of these genes appeared to be expressed specifically in pollen, similar to the situation in the S locus in P. dulcis and P. mume (Entani et al. 2003; Ushijima et al. 2003). This may simply be fortuitous for the S 2 allele because much larger physical separations occur between S-RNase and AhSLF gene clusters in the other three alleles (Figs. 1, 3). AhSLF-S 2 represents the closest gene to an S-RNase identified so far in Antirrhinum. We also compared the putative amino acid sequences of AhSLF-S and SLF genes from P. dulcis and P. mume (see Table 2), but the similarity between them was very low (15–25%). Thus, it is difficult to conclude which kind of F-box gene in almond is more similar to AhSLF-S. So far, we have not found SLF genes with high polymorphism in Antirrhinum. There are two possibilities: either there are no high polymorphic SLF genes in Antirrhinum or, different from the two rosaceous species, such polymorphic SLF are located further from the S-RNases. Further work to close the gaps between S-RNase-containing TAC clones and AhSLF regions in other three S haplotypes may help resolve this point in Antirrhinum.
Intriguingly, it is not clear whether AhSLF-S genes with such high identity to each other are capable of encoding Sp. Recently, Luu et al. (2001) have proposed a model for Sp consisting of two components, a general S-RNase inhibitor (RI) capable of inhibiting any S-RNase, and an S allele-specific product that maintains the activity of a specific S-RNase inside the pollen tube by blocking RI binding. Whether AhSLF-S genes could encode such a general S-RNase inhibitor awaits further investigation. However, AhSLF-S is different from a recently described petunia protein (PhSBP1) that interacts with S-RNase (Sims and Ordanic 2001) because the latter appears to be expressed ubiquitously.
In addition, several SLF genes are also detected near the S 2 -RNase gene. The absence of expression of AhSLF-SA alleles in pollen ruled out their role in the self-incompatible reaction. However, it is unclear what roles other pollen-specific SLF genes could play. It is possible that the paralogous AhSLF genes represent ancient duplications of the AhSLF-S gene that occurred during early angiosperm diversification, leaving other AhSLF genes near the border of the S locus but playing no role in SI response. However, it is not clear why the linkage to the S locus is needed if AhSLF-S products indeed act as general RNase inhibitors. It would make sense if they simply played a general role in other unknown aspects of pollen growth, their location near the S-RNase genes being simply fortuitous. As a result, the observed diversity of AhSLF genes would have occurred because of a "hitchhiking" effect due to suppressed recombination in the S locus region. Another possibility is that the association of S-RNase and AhSLF-S derives from a pre-existing linkage of ancestral S-like RNase and SLF genes during earlier angiosperm formation. As discussed by Luu et al. (2000), RNase-based SI could be derived through a step-wise process. In this scenario, a pre-existing ancestral linkage between an inhibitor and an S-like RNase was maintained to allow inhibition—and thus pollen survival—because of the free-entry of S RNases into the pollen tube. Eventually, a factor (presumably Sp) attenuating or abolishing this inhibition would have been selected for due to the advantage of self-pollen rejection. Transgenic experiments to test the function of the AhSLF-S 2 gene in Antirrhinum together with detailed yeast two-hybrid screening and biochemical studies on AhSLF-S are clearly required to address these issues.
References
Ai Y, Singh A, Coleman CE, Ioerger TR, Kheyr-Pour A (1990) Self-incompatibility in Petunia inflata: isolation and characterization of cDNA encoding S-allele-associated proteins. Sex Plant Reprod 3:130–138
Anderson MA, Cornish EC, Mau SL, Williams EG, Hoggart R, Atkinson A, Bonig I, Grego B, Simpson R, Roche PJ, Haley JD, Penshow JD, Niall HD, Tregear GW, Coghlan JP, Crawford RJ, Clarke AE (1986) Cloning of a cDNA for a style glycoprotein associated with expression of self-incompatibility in Nicotiana alata. Nature 321:38–44
Broothaerts W, Janssens GA, Proost P, Broekaert W (1995) cDNA cloning and molecular analysis of two self-incompatibility alleles from apple. Plant Mol Biol 27:499–511
Cheng Z, Dong F, Langdon T, Ouyang S, Buell CR (2002) Functional rice centromeres are marked by a satellite repeat and a centromere-specific retrotransposon. Plant Cell 14:1691–1704
Coleman CE, Kao TH (1992) The flanking region of two Petunia inflate S alleles are heterogenous and contain repetitive sequences. Plant Mol Biol 18:725–737
Copenhaver GP, Nickel K, Kuromori T, Benito MI, Kaul S (1999) Genetic definition and sequence analysis of Arabidopsis centromeres. Science 286:2468–2474
Duret L, Marais G, Biemont C (2000) Transposons but not retrotransposons are located preferentially in regions of high recombination rate in Caenorhabditis elegans. Genetics 156:1661–1669
Ellis J, Dodds P, Pryor T (2000a) The generation of plant disease resistance gene specificities. Trends Plant Sci 5:373–379
Ellis J, Dodds P, Pryor T (2000b) Structure, function and evolution of plant disease resistance genes. Curr Opin Plant Biol 3:278–284
Emerson S (1939) A preliminary survey of the Oenothera organensis population. Evolution 24:524–537
Entani T, Iwano M, Shiba H, Takayama S, Fukui K (1999) Centromeric localization of an S-RNase gene in Petunia hybrida Vilm. Theor Appl Genet 99:391–397
Entani T, Iwano M, Shiba H, Che F, Isogai A, Takayama S (2003) Comparative analysis of the self-incompatibility (S-) locus region of Prunus mume: identification of a pollen-expressed F-box gene with allelic diversity. Genes Cells 8:203–213
Foote HC, Ride JP, Franklin-Tong VE, Walker EA, Lawrence MJ, Franklin FCH (1994) Cloning and expression of a distinctive class of self-incompatibility (S-) gene from Papaver rhoeas L. Proc Natl Acad Sci USA 91:2265–2269
Frey M, Tavantzis SM, Saedler H (1989) The maize En/Spm element transposes in tomato. Mol Gen Genet 217:172–177
Fu H, Zheng Z, Dooner HK (2002) Recombination rates between adjacent genic and retrotransposon region in maize vary by 2 orders of magnitude. Proc Natl Acad Sci USA 99:1082–1087
Golz JF, Su V, Clarke AE, Newbigin EA (1999) A molecular description of mutations affecting the pollen component of the Nicotiana alata S locus. Genetics 152:1123–1135
Golz JF, Clarke AE, Newbegin E (2000) Mutational approaches to the study of self-incompatibility: revisiting the pollen part mutations. Ann Bot 85:95–103
Golz JF, Oh HY, Su V, Kusaba M, Newbigin E (2001) Genetics analysis of Nicotiana pollen-part mutant is consistent with the presence of an S-ribonuclease inhibitor at the S locus. Proc Natl Acad Sci USA 98:15372–15376
Hammond-Kosack KE, Tang S, Harrison K, Jones JD (1998) The tomato Cf-9 disease resistance gene functions in tobacco and potato to confer responsiveness to the fungal avirulence gene product Avr 9. Plant Cell 10:1251–1266
Igic B, Kohn JR (2001) Evolutionary relationships among self-incompatiblity RNases. Proc Natl Acad Sci USA 98:13167–13171
Kachroo A, Schopfer CR, Nasrallah ME, Nasrallah JB (2001) Allele-specific receptor-ligand interactions in Brassica self-incompatibility. Science 293 1824–1826
Lai Z, Ma W, Han B, Liang L, Zhang Y, Hong G, Xue Y (2002) An F-box gene linked to the self-incompatibility (S) locus of Antirrhinum is expressed specifically in pollen and tapetum. Plant Mol Biol 50:29–42
Lawrence MJ, Lane MD, O'Donnell S, Franklin-Tong VE (1993) The population genetics of the self-incompatibility polymorphism in Papaver rhoeas. V. Cross-classification of the S-alleles of samples from three natural populations. Heredity 71:581–590
Li JH, Nass N, Kusaba M, Dodds PN, Treloar N, Clarke AE, Newbigin E (2000) A genetic map of the Nicotiana alata S locus that includes three pollen-expressed genes. Theor Appl Genet 100:956–964
Liu YG, Whitter RF (1994) Rapid preparation of megabase plant DNA from nuclei in agarose plugs and microbeads. Nucleic Acids Res 22:2168–2169
Liu YG, Shirano Y, Fukaki H, Yanai Y, Tasaka M (1999) Complementation of plant mutants with large genomic DNA fragments by a transformation-competent artificial chromosome vector accelerates positional cloning. Proc Natl Acad Sci USA 96:6535–6540
Liu YG, Nagaki K, Fujita M, Kawaura K, Uozumi M, Ogihara Y (2000) Development of an efficient maintenance and screening system for large-insert genomic DNA libraries of hexaploid wheat in transformation-competent artificial chromosome (TAC) vector. Plant J 23:687–695
Luu DT, Qin X, Morse D, Cappadocia M (2000) S-RNase uptake by compatible pollen tubes in gametophytic self-incompatibility. Nature 407:649–651
Luu DT, Qin X, Laublin G, Yang Q, Morse D, Cappadocia M (2001) Rejection of S-heteroallelic pollen by a dual-specific S-RNase in Solanum chacoense predicts a multimeric SI pollen component. Genetics 159:329–335
Ma W, Zhou J, Lai Z, Zhang Y, Xue Y (2002) The self-incompatibility (S) locus is located in a pericentromeric region in Antirrhinum. Acta Bot Sin 45:47–52
Martin GB, Brommonschenkel SH, Chunwongse J, Frary A, Ganal MW (1993) Map-based cloning of a protein kinase gene conferring disease resistance in tomato. Science 262:1432–1436
May G, Matzke E (1995) Recombination and variation at the mating-type of Coprinus-cinereus. Mol Biol Evol 12:794–802
McClure BA, Haring V, Ebert PR, Anderson MA, Simpson RJ, Sakiyama F, Clarke AE (1989) Style self-incompatibility gene products of Nicotiana alata are ribonucleases. Nature 342:955–957
McCubbin AG, Kao TH (2000) Molecular recognition and response in pollen and pistil interactions. Annu Rev Cell Dev Biol 16:333–364
McCubbin AG, Wang X, Kao TH (2000) Identification of self-incompatibility (S-) locus linked pollen cDNA markers in Petunia inflata. Genome 43:619–627
Michelmore RW, Meyers BC, (1998) Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res 8:1113–1130
Nacken WK, Piotrowiak R, Suedler H, Sommer H (1991) The transposable element Tam1 from Antirrhinum majus shows structural homology to the maize transposon En/Spm and has no sequence specificity of insertion. Mol Gen Genet 228:201–208
Nettancourt D de (2001) Incompatibility and incongruity in wild and cultivated plants, 2nd edn. Springer, Berlin Heidelberg New York
O'hUigin C (1995) Quantifying the degree of convergence in primate MHC-DRB genes. Immunol Rev 143:123–140
Pandy KK (1965) Centric chromosome fragments and pollen-part mutation of the incompatibility gene in Nicotiana alata. Nature 206:792–795
Richman AD, Uyenoyama MK, Kohn JR (1996) S-allele diversity in a natural population of Physalis crassifolia (Solanaceae) (ground cherry) assessed by RT-PCR. Heredity 76:497–505
Rizzon C, Marais G, Gouy M, Biemont C (2002) Recombination rate and the distribution of transposable elements in the Drosophila melanogaster genome. Genome Res 12:400–407
Royo J, Nass N, Matton DP, Okamto S, Clarke AE, Newbigin E (1996) A retrotransposon-like sequence linked to the S-locus of Nicotiana alata is expressed in styles in response to touch. Mol Gen Genet 250:180–188
Sassa H, Nishio T, Kowyama Y, Hirano H, Koba T (1996) Self-incompatibility (S) alleles of the Rosaceae encode members of a distinct class of the T2/S ribonuclease superfamily. Mol Gen Genet 250:547–557
Schopfer CR, Nasrallah ME, Nasrallah JB (1999) The male determinant of self-incompatibility in Brassica. Science 286:1697–1700
Sims TL, Ordanic M (2001) Identification of a S-ribonuclease-binding protein in Petunia hybrida. Plant Mol Biol 47:771–783
Stein JC, Howlett B, Boyes DC, Nasrallah ME, Nasrallah JB (1991) Molecular cloning of a putative receptor protein kinase encoded at the self-incompatibility locus of Brassica oleracea. Proc Natl Acad Sci USA 88:8816–8820
Steinbachs JE, Holsinger KE (2002) S-RNase-mediated gametophytic self-incompatibility is ancestral in eudicots. Mol Biol Evol 19:825–829
Suzuki G, Kai N, Hirose T, Fukui K, Nishio T (1999) Genomic organization of the S locus: Identification and characterization of genes in SLG/SRK region of S(9) haplotype of Brassica campestris(syn. rapa). Genetics 3:391–400
Takayama S, Shimosato H, Shiba H, Funata M, Che FS, Watanabe M, Iwano M, Isogai A (2001) Direct ligand-receptor complex interaction controls Brassica self-incompatibility. Nature 413:534–536
Ushijima K, Sassa H, Hirano H (1998) Characterization of the flanking regions of S-RNase genes of Japanese pear (Pyrus serotina) and apple (Malus×domestica). Gene 211:159–167
Ushijima K, Sassa H, Dandekar A, Gradziel T, Tao R, Hirano H (2003) Structure and transcriptional analysis of the self-incompatibility locus of Almond: identification of a pollen-expressed F-box gene with haplotype-specific polymorphism. Plant Cell 15:771–781
Wei F, Wing RA, Wise RP (2002) Genome dynamics and evolution of the Mla (Powdery Mildew) resistance locus in barley. Plant Cell 14:1903–1917
Wheeler MJ, Franklin-Tong VE, Franklin FCH (2001) The molecular and genetics basis of pollen-pistil interactions. New Phytol 151:565–584
Xue Y, Carpenter R, Dickinson HG, Coen ES (1996) Origin of allelic diversity in Antirrhinum S locus RNases. Plant Cell 8:805–814
Acknowledgements
We are grateful to E.S. Coen and R. Carpenter for providing Antirrhinum plants and constant support and Y. Liu for help in constructing TAC library. We also thank J. Huang, H. Qiao and H. Wang and, in particular, an anonymous reviewer for their helpful comments on the manuscript. The work was supported by the Chinese Academy of Sciences and the National Natural Science Foundation of China (39825103).
Author information
Authors and Affiliations
Corresponding author
Additional information
Sequence data from this article have been deposited with the EMBL/GenBank databases under accession numbers AJ300474, AJ515534, AJ515536 and AJ515535
Rights and permissions
About this article
Cite this article
Zhou, J., Wang, F., Ma, W. et al. Structural and transcriptional analysis of S-locus F-box genes in Antirrhinum . Sex Plant Reprod 16, 165–177 (2003). https://doi.org/10.1007/s00497-003-0185-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00497-003-0185-5