
Abstract
Background
Heterozygous PAX2 mutations cause renal coloboma syndrome (RCS) [OMIM no. 120330]. RCS is a renal syndromic disease encompassing retinal coloboma and sensorineural hearing loss. Recently, a causative role for PAX2 was reported in adult-onset nephrotic syndrome secondary to focal segmental glomerulosclerosis (FSGS). However, the prevalence of PAX2 mutations among large cohort of children with steroid-resistant nephrotic syndrome (SRNS) and FSGS has not been systematically studied.
Methods
We employed whole-exome sequencing (WES) to identify the percentage of SRNS cases explained by monogenic mutations in known genes of SRNS/FSGS. As PAX2 mutations are not an established cause of childhood FSGS, we evaluated a cohort of 215 unrelated families with SRNS, in whom no underlying genetic etiology had been previously established.
Results
Using WES, we identified 3 novel causative heterozygous PAX2 mutations in 3 out of the 215 unrelated index cases studied (1.3%). All three cases were detected in individuals from families with more than one affected and compatible with an autosomal dominant mode of inheritance (3/57 familial cases studied (5.2%)). The clinical diagnosis in three out of four pediatric index patients was done during routine medical evaluation.
Conclusions
Our findings demonstrate high frequency of PAX2 mutations in familial form of SRNS (5.2%) and further expand the phenotypic spectrum of PAX2 heterozygous mutations to include autosomal dominant childhood-onset FSGS. These results highlight the importance of including PAX2 in the list of genes known to cause FSGS in children.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
PAX2 (Paired Box gene 2) is a transcription factor that plays a central role during early embryonic kidney development. Heterozygous mutations in PAX2 were first identified in patients with renal coloboma syndrome (also known as “papillorenal syndrome”) which involves renal hypodysplasia, optic nerve abnormalities, and deafness [1]. Nonetheless, PAX2 heterozygous mutations can lead to variable kidney phenotypes across the morphologic continuum of CAKUT (congenital anomalies of the kidney and urinary tract) [2]. This includes renal hypodysplasia, renal cysts, multicystic dysplastic kidneys, and vesicoureteral reflux as the most common ones [3]. Furthermore, pathogenic mutations can lead to isolated CAKUT without optic nerve or hearing abnormalities or with only subtle features of CAKUT [2] [4].
In the absence of a CAKUT phenotype, there is some data suggesting a causative role for PAX2 mutations among individuals with nephrotic syndrome or nephrotic range proteinuria and focal segmental glomerulosclerosis (FSGS) [5, 6]. These observations have been documented exclusively for adult-onset nephrotic syndrome secondary to FSGS [5]; however, the prevalence of PAX2 mutations among large cohort of children with steroid-resistant nephrotic syndrome (SRNS) and FSGS has not been systematically studied.
In the present study, following identification of a child with coloboma, nephrotic range proteinuria and renal histology showing FSGS, and in whom we detected a de novo pathogenic mutation in PAX2, we hypothesized that PAX2 mutations may be relatively prevalent among children diagnosed with steroid-resistant nephrotic syndrome/FSGS and who have no molecular genetic diagnosis. Accordingly, we analyzed whole-exome sequencing data from 215 different index cases with steroid-resistant nephrotic syndrome in which no causative mutation in a known SRNS gene has been previously identified [7,8,9]. In 57 families, there was more than one affected individual. Using this strategy, we identified PAX2 mutations in 3 out of the 215 index cases studied (1.3%). All 3 cases were detected in families with more than one affected individual (3/57 familial cases (5.2%)). In all three cases, we established a specific etiologic genetic diagnosis that was not made on the basis of the patient’s clinical findings.
Methods
Study participants
The study was approved by the Institutional Review Board of the University of Michigan and Boston Children’s Hospital. From April of 1998 to June of 2016, patients were enrolled after obtaining informed consent [10]. Inclusion criteria were onset of symptoms before 25 years and a clinical diagnosis of steroid-resistant nephrotic syndrome (e.g., proteinuria, hypoalbuminemia, and edema) or nephrotic range proteinuria with kidney histology of FSGS or diffuse mesangial sclerosis [10]. Excluded from the study were patients with one of the following: (1) non-nephrotic range proteinuria or isolated hematuria; (2) patients with steroid-sensitive nephrotic syndrome, (3) patients with steroid-dependent nephrotic syndrome, (4) patients with acute glomerulonephritis; (5) patients older than 25 years at nephrotic syndrome onset, and (6) patients with known monogenic etiology. Mutations in the following 32 genes known to be mutated in steroid-resistant nephrotic syndrome were excluded by whole-exome sequencing (WES) [10] before this study: ACTN4, ADCK4, ARHGDIA, CD2AP, COQ2, COQ6, CRB2, CUBN, DGKE, FAT1, INF2, ITGA3, KANK1, KANK2, KANK4, LAMB2, LMX1B, MYO1E, NPHS1, NPHS2, NUP205, NUP93, PDSS2, PLCE1, SGPL1, SMARCAL1, TRPC6, TTC21B, WDR73, WT1, XPO5, AGXT, COL4A3, COL4A4, COL4A5, CLCN5, CTNS, FN1, GLA, LRP2, MEVF, and OCRL.
Description of cohort
Our original cohort included 300 index cases with SRNS. Onset of SRNS in our cohort ranged from birth to 24 years of age. Approximately 55% of cases had onset of SRNS before 6 years of age [10]. We examined each family’s pedigree for consanguinity and evaluated WES data for homozygosity as well as for 1 of the 32 SRNS-causing genes noted above. We previously identified known SRNS-causing genes in 74 families and mutations in genes causing SRNS phenocopy in 11 families [10]. These families were excluded from the current study. We reviewed each individual for extra-renal manifestations, including cardiac defects, microcephaly, and limb abnormalities. Twenty-seven percent of patients in our cohort had extra-renal manifestations. The cohort’s most common clinical diagnosis was SRNS in two thirds of the cohort compared to those who were diagnosed with congenital nephrotic syndrome, infantile nephrotic syndrome, or those with biopsy evidence of FSGS or DMS (diffuse mesangial sclerosis) but were not clinically nephrotic [10]. Overall, our cohort included clinical data as well as WES data from a total of 215 individuals from different families with steroid resistance nephrotic syndrome without any known monogenic etiology.
Whole-exome sequencing and variant calling
Whole-exome sequencing (WES) was performed using genomic DNA isolated from blood lymphocytes and later processed using Agilent SureSelect human exome capture arrays (Life Technologies™) with next-generation sequencing on an Illumina™ sequencing platform at the Broad Institute (Cambridge, MA) and Yale Center for Mendelian Genomics (New Haven, CT). Sequence reads were mapped to the human reference genome assembly (NCBI build 37/hg19 www.genome.ucsc.edu) using CLC Genomics Workbench (version 6.5.1) software (CLC bio, Aarhus, Denmark) as previously described [11]. Mutation calling for known steroid-resistant nephrotic syndrome genes has been previously described [10]. As noted above, these families were excluded from the current study. WES Trio analysis for the index family was performed as previously described [12].
PAX2 variant calling
Following WES, PAX2 genetic variants were first filtered to retain only non-synonymous and splice variants. Second, filtering was performed to retain only alleles with a minor allele frequency (MAF) of zero. MAF was estimated using combined datasets incorporating all available data from the 1000 Genomes Project, the Exome Variant Server (EVS) project, dbSNP142, and the Exome Aggregation Consortium (ExAC). Third, observed sequence variants were analyzed using the UCSC Human Genome Bioinformatics Browser for the presence of paralogous genes, pseudogenes, or misalignments. Fourth, we scrutinized all variants within the sequence alignments of the CLC Genomic Workbench™ software program for poor sequence quality and for the presence of mismatches that indicate potential false alignments. Fifth, we employed web-based programs to assess variants for evolutionary conservation, to predict the impact of disease candidate variants on the encoded protein, and to determine whether these variants represented known disease-causing mutations. All bioinformatics analysis was performed by clinician scientists, with knowledge of the clinical phenotypes and pedigree structure, as well as experience with whole-exome sequencing evaluation. Sanger sequencing was performed to confirm the remaining variants in original DNA samples and when available to test for familial segregation of phenotype with genotype.
Results
WES trio analysis identifies an index case of FSGS and PAX2 mutation
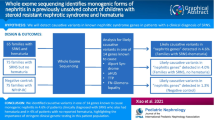
We consulted the index family (AN10), a non-consanguineous Bedouin pedigree (Fig. 1). The index patient was a female born via cesarean section at term. Prenatal ultrasound examination during the second trimester demonstrated microcephaly and mild cardiac hypertrophy. Following birth, the patient was diagnosed with mild hypertrophic cardiomyopathy and later developed hypertension. She started treatment with a non-selective beta blocker/alpha-1 blocker (Carvedilol) and has been well controlled. Post-natal metabolic work-up including urine organic acids, acyl-carnitine profile, plasma carnitine, and lactate was unremarkable. In addition, muscle biopsy was normal. Consequently, during infancy, her renal function gradually deteriorated (serum creatinine level at age 1 year was 0.8–0.9 mg/dL [normal range 0.2–0.5 mg/day]) and she developed nephrotic range proteinuria with urine protein to creatinine ratio (UPC) of 6.7 mg/mg. Renal US demonstrated mildly echogenic kidneys with normal shape and size, without evidence of hydronephrosis or bladder abnormalities. Subsequent renal biopsy demonstrated focal segmental glomerulosclerosis (FSGS) and no features of renal hypodysplasia (Fig. 1). The patient did not respond to steroid treatment and subsequently developed end-stage renal disease. She required renal replacement therapy at the age of 4 years and received a cadaveric renal transplant at the age of 6 years. At the same time, a routine eye examination revealed bilateral coloboma of the optic nerve.

Pedigree structures and representative renal histology findings in patients with steroid-resistant nephrotic syndrome and PAX2 mutations. a Pedigree structure of index family (AN_10) and three families with FSGS and PAX2 mutations (A4041, A5089, and A5281). b Pathologic findings in renal biopsy specimen from affected individual in family AN_10 showing segmental glomerulosclerosis (black arrow head). c Pathologic findings in renal biopsy specimen from affected proband in family A4041 showing segmental glomerulosclerosis (black arrow head). Bx, Biopsy; ESRD, end-stage renal disease; N/A, not available; ND, not determent
For this family, we performed trio WES analysis. First, we analyzed all known nephrotic syndrome-causing genes; this analysis was negative. Next, we performed analysis under autosomal recessive hypothesis—which also yielded negative results. Finally, we performed de novo analysis and detected a previously reported heterozygous truncating mutation in PAX2 (c.69-70insG; p.Val26Glyfs*28). We [13] as well as others [14,15,16] showed that this particular truncating mutation resides in a mutation hot spot region and for which a germline mosaicism inheritance pattern has been suggested previously in three different families [14,15,16]. Interestingly, previous mouse model carrying this mutation [17] showed abnormal brain development, in addition to the renal, eye, and kidney phenotype. This may link the patient’s microcephaly to the underlying PAX2 mutation.
Identification of causative mutations in PAX2 in three families with steroid-resistant nephrotic syndrome and FSGS
We next analyzed the prevalence of PAX2 mutations in a cohort of 215 index cases with steroid-resistant nephrotic syndrome. Surprisingly, in three families with autosomal dominant familial form of SRNS (Fig. 1), we identified three different novel PAX2 mutations. This included two missense mutations affecting a highly conserved amino acid (up to Danio rerio) and one obligatory splice mutation. In each family, the PAX2 mutation co-segregated across all affected available to us (Table 1). Overall, we identified PAX2 mutations in 3 out of the 215 index cases studied (1.3%). All 3 cases were detected in families with more than one affected (3/57 familial cases (5.2%)).
In family A4041, we detected a novel missense mutation affecting the PAX2 protein paired domain (c.254G > T; p.Gly85Val). In this family, two affected siblings presented during adolescence with nephrotic range proteinuria and impaired GFR. Renal biopsy findings for both siblings showed FSGS (Fig. 1). Both failed to respond to steroid treatment. Their father was transplanted at age 40 years but presented with nephrotic range proteinuria and advanced chronic kidney disease at age 35 years. All three affected family members available to us carried the heterozygous PAX2 mutation. All affected had no overt coloboma, hearing impairment, or other extra-renal or syndromic features. Renal ultrasound studies as well as renal histology showed no signs of CAKUT.
In family A5089, we detected a novel PAX2 splice mutation affecting an obligatory splice site, predicted to lead to skip of exon 8 (c.862-1G > A). In this family, the index child presented during adolescence with nephrotic range proteinuria (UPC = 8), elevated creatinine, and low albumin, which were found incidentally. Renal biopsy showed FSGS. On presentation, the patient had severely depressed GFR of 16 ml/min/1.73m2 and subsequently required renal replacement therapy. As a result, she was not treated with steroids. The patient’s father had, at the age of 55 years, normal renal function and nephrotic range proteinuria. The affected had neither extra-renal nor syndromic features nor renal ultrasound or histology features of CAKUT.
In family A5281, we detected a novel missense mutation affecting the PAX2 protein paired domain (c.275G > T; p.Thr92Met). In this family, the index patient presented incidentally with hypertension which was diagnosed on routine examination. He was found to have nephrotic range proteinuria and renal US which did not show CAKUT. His renal biopsy was remarkable for FSGS. Interestingly, his mother as well as his maternal grandfather had end-stage renal disease and required renal replacement therapy during adulthood. Unfortunately, for both, DNA and renal biopsy findings were not available to us.
Discussion
In the current study, we examined a group of 215 unrelated index cases with steroid resistance nephrotic syndrome for the presence of heterozygous PAX2 disease-causing mutations. We identified three different novel PAX2 mutations which co-segregated across all affected individuals available to the study (family A4041 and A5089). All variants were novel and were predicted to be deleterious (Table 1). Specifically, two variants affect the PAX2 protein paired domain and one affect an obligatory splice site leading to exon skipping. In all patients, CAKUT phenotype was excluded as well as disease-causing mutations in known SRNS genes. None of the PAX2 disease-causing mutations was suspected on clinical grounds prior to the current study, and affected patients were not clinically distinguished from other steroid-resistant nephrotic syndrome patients. Our study highlights several important conclusions: First, we expanded the phenotypic spectrum associated with PAX2 mutations and further established it to include autosomal dominant childhood-onset FSGS. It also further demonstrates the variable expressivity related to PAX2 mutations as the phenotypical severity and presentation in our cohort was variable. Second, utilizing a large cohort of patients with SRNS allowed us to determine PAX2 mutation frequency (5.2% in multiplex families with SRNS). Third, the diagnosis of PAX2-related SRNS may inform clinical practice as those patients fail to respond to steroid treatment. In addition, it may trigger clinician to look for subtle extra-renal findings such as coloboma or hearing impairment. Fourth, in three out of the fourth index children studied, the clinical diagnosis was made incidentally. This can explain previous observation ascribing PAX2 mutations to adult-onset FSGS. It also emphasizes the importance of urinalyses screening programs for children. Finally, this study highlights the potential benefits of clarifying diagnosis in SRNS through WES to avoid unnecessary invasive procedures and treatments, especially given that PAX2 mutations may account for a significant proportion of families with autosomal dominant form of SRNS.
Pax genes are among the many types of prototypical DNA-binding proteins important for cell lineage specification in multicellular organisms [18]. In mice and humans, Pax2 genes encode proteins with amino terminal DNA binding, i.e., paired domains. Several paired domain PAX2 mutations related to adult-onset FSGS have been reported; however, no genotype phenotype correlation has been observed to date [5].
There are several possible explanations for the mechanisms relating PAX2 mutations and glomerular injury such as FSGS. First, during kidney development, PAX2 is important in order to allow for proper communication between the ureteric bud and surrounding metanephric mesenchyme, the latter of which ultimately epithelializes to form podocytes. Specifically, it has been recently shown that nephron progenitor cells lacking Pax2 fail to differentiate into nephron cells. These data suggest that Pax2 function maintains nephron progenitor cells and hence may affect the final function of the glomeruli [19]. Second, previous data suggests that PAX2 strongly represses the expression of WT1 and vice versa. WT1 is a nuclear protein expressed in podocytes [20]. This dysregulation of PAX2 targets, such as WT1, may lead to FSGS by disrupting the development and/or function of the podocyte. Finally, the Pax-2 transgenic mice phenotype, reported by Dressler et al. [21], exclusively exhibited significant proteinuria. In addition, renal ultrastructural analysis of those mice showed paucity of podocyte foot processes and poorly developed endothelial fenestrae [21].
In summary, we identified PAX2 mutations in 3 out of the 215 index cases studied (1.3%). All three cases were detected in families with more than one affected individual with SRNS/FSGS (3/57 families (5.2%)). In all three cases, we established a specific etiologic genetic diagnosis that was not made on the basis of the patient’s clinical findings. Our results further establish the causative role of PAX2 among children with SRNS/FSGS.
References
Sanyanusin P, Schimmenti LA, McNoe LA, Ward TA, Pierpont ME, Sullivan MJ, Dobyns WB, Eccles MR (1995) Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat Genet 9:358–364
Weber S, Moriniere V, Knuppel T, Charbit M, Dusek J, Ghiggeri GM, Jankauskiene A, Mir S, Montini G, Peco-Antic A, Wuhl E, Zurowska AM, Mehls O, Antignac C, Schaefer F, Salomon R (2006) Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol: JASN 17:2864–2870
Bower M, Salomon R, Allanson J, Antignac C, Benedicenti F, Benetti E, Binenbaum G, Jensen UB, Cochat P, DeCramer S, Dixon J, Drouin R, Falk MJ, Feret H, Gise R, Hunter A, Johnson K, Kumar R, Lavocat MP, Martin L, Moriniere V, Mowat D, Murer L, Nguyen HT, Peretz-Amit G, Pierce E, Place E, Rodig N, Salerno A, Sastry S, Sato T, Sayer JA, Schaafsma GC, Shoemaker L, Stockton DW, Tan WH, Tenconi R, Vanhille P, Vats A, Wang X, Warman B, Weleber RG, White SM, Wilson-Brackett C, Zand DJ, Eccles M, Schimmenti LA, Heidet L (2012) Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus-specific database. Hum Mutat 33:457–466
Bower MA, Schimmenti LA, Eccles MR (1993) PAX2-Related Disorder. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (eds) GeneReviews((R)), Seattle (WA)
Barua M, Stellacci E, Stella L, Weins A, Genovese G, Muto V, Caputo V, Toka HR, Charoonratana VT, Tartaglia M, Pollak MR (2014) Mutations in PAX2 associate with adult-onset FSGS. J Am Soc Nephrol: JASN 25:1942–1953
Okumura T, Furuichi K, Higashide T, Sakurai M, Hashimoto S, Shinozaki Y, Hara A, Iwata Y, Sakai N, Sugiyama K, Kaneko S, Wada T (2015) Association of PAX2 and other gene mutations with the clinical manifestations of renal coloboma syndrome. PLoS One 10:e0142843
Vivante A, Hildebrandt F (2016) Exploring the genetic basis of early-onset chronic kidney disease. Nat Rev Nephrol 12:133–146
Lovric S, Ashraf S, Tan W, Hildebrandt F (2016) Genetic testing in steroid-resistant nephrotic syndrome: when and how? Nephrol Dial Transplant 31(11):1802–1813
Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, Engelmann S, Vega-Warner V, Fang H, Halbritter J, Somers MJ, Tan W, Shril S, Fessi I, Lifton RP, Bockenhauer D, El-Desoky S, Kari JA, Zenker M, Kemper MJ, Mueller D, Fathy HM, Soliman NA, Hildebrandt F (2015) A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol: JASN 26:1279–1289
Warejko JK, Tan W, Daga A, Schapiro D, Lawson JA, Shril S, Lovric S, Ashraf S, Rao J, Hermle T, Jobst-Schwan T, Widmeier E, Majmundar AJ, Schneider R, Gee HY, Schmidt JM, Vivante A, van der Ven AT, Ityel H, Chen J, Sadowski CE, Kohl S, Pabst WL, Nakayama M, Somers MJG, Rodig NM, Daouk G, Baum M, Stein DR, Ferguson MA, Traum AZ, Soliman NA, Kari JA, El Desoky S, Fathy H, Zenker M, Bakkaloglu SA, Muller D, Noyan A, Ozaltin F, Cadnapaphornchai MA, Hashmi S, Hopcian J, Kopp JB, Benador N, Bockenhauer D, Bogdanovic R, Stajic N, Chernin G, Ettenger R, Fehrenbach H, Kemper M, Munarriz RL, Podracka L, Buscher R, Serdaroglu E, Tasic V, Mane S, Lifton RP, Braun DA, Hildebrandt F (2018) Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol : CJASN 13:53–62
Vivante A, Hwang DY, Kohl S, Chen J, Shril S, Schulz J, van der Ven A, Daouk G, Soliman NA, Kumar AS, Senguttuvan P, Kehinde EO, Tasic V, Hildebrandt F (2017) Exome sequencing discerns syndromes in patients from consanguineous families with congenital anomalies of the kidneys and urinary tract. J Am Soc Nephrol 28:69–75
Vivante A, Ityel H, Pode-Shakked B, Chen J, Shril S, van der Ven AT, Mann N, Schmidt JM, Segel R, Aran A, Zeharia A, Staretz-Chacham O, Bar-Yosef O, Raas-Rothschild A, Landau YE, Lifton RP, Anikster Y, Hildebrandt F (2017) Exome sequencing in Jewish and Arab patients with rhabdomyolysis reveals single-gene etiology in 43% of cases. Pediatr Nephrol 32:2273–2282
Vivante A, Mark-Danieli M, Davidovits M, Harari-Steinberg O, Omer D, Gnatek Y, Cleper R, Landau D, Kovalski Y, Weissman I, Eisenstein I, Soudack M, Wolf HR, Issler N, Lotan D, Anikster Y, Dekel B (2013) Renal hypodysplasia associates with a WNT4 variant that causes aberrant canonical WNT signaling. J Am Soc Nephrol 24:550–558
Sanyanusin P, McNoe LA, Sullivan MJ, Weaver RG, Eccles MR (1995) Mutation of PAX2 in two siblings with renal-coloboma syndrome. Hum Mol Genet 4:2183–2184
Cheong HI, Cho HY, Kim JH, Yu YS, Ha IS, Choi Y (2007) A clinico-genetic study of renal coloboma syndrome in children. Pediatr Nephrol 22:1283–1289
Amiel J, Audollent S, Joly D, Dureau P, Salomon R, Tellier AL, Auge J, Bouissou F, Antignac C, Gubler MC, Eccles MR, Munnich A, Vekemans M, Lyonnet S, Attie-Bitach T (2000) PAX2 mutations in renal-coloboma syndrome: mutational hotspot and germline mosaicism. Eur J Hum Genet : EJHG 8:820–826
Favor J, Sandulache R, Neuhauser-Klaus A, Pretsch W, Chatterjee B, Senft E, Wurst W, Blanquet V, Grimes P, Sporle R, Schughart K (1996) The mouse Pax2(1Neu) mutation is identical to a human PAX2 mutation in a family with renal-coloboma syndrome and results in developmental defects of the brain, ear, eye, and kidney. Proc Natl Acad Sci U S A 93:13870–13875
Dressler GR (2011) Patterning and early cell lineage decisions in the developing kidney: the role of Pax genes. Pediatr Nephrol 26:1387–1394
Naiman N, Fujioka K, Fujino M, Valerius MT, Potter SS, McMahon AP, Kobayashi A (2017) Repression of interstitial identity in nephron progenitor cells by Pax2 establishes the nephron-Interstitium boundary during kidney development. Dev Cell 41:349–365 e343
Wagner KD, Wagner N, Guo JK, Elger M, Dallman MJ, Bugeon L, Schedl A (2006) An inducible mouse model for PAX2-dependent glomerular disease: insights into a complex pathogenesis. Curr Biol 16:793–800
Dressler GR, Wilkinson JE, Rothenpieler UW, Patterson LT, Williams-Simons L, Westphal H (1993) Deregulation of Pax-2 expression in transgenic mice generates severe kidney abnormalities. Nature 362:65–67
Funding
F.H. the William E. Harmon Professor of Pediatrics was supported by a grant from the National Institutes of Health to FH (R01-DK076683). A.V. is a recipient of the Fulbright Post-doctoral Scholar Award for 2013. A.V. is also supported by grants from the Manton Center Fellowship program, Boston Children’s Hospital, Boston, MA and the Mallinckrodt Research Fellowship Award. This work was also supported by German Federal Ministry for Education and Research (BMBF) Grant STOP-FSGS 01GM1518A (to M. S.). Sequencing and data processing was performed by the Broad and Yale Centers for Mendelian Genomics funded by the National Human Genome Research Institute (UM1 HG008900 to DGM and HLR and U54 HG006504 to RPL).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study was approved by the Institutional Review Board of the University of Michigan and Boston Children’s Hospital.
Conflict of interest
The authors declare that there are no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Vivante, A., Chacham, O.S., Shril, S. et al. Dominant PAX2 mutations may cause steroid-resistant nephrotic syndrome and FSGS in children. Pediatr Nephrol 34, 1607–1613 (2019). https://doi.org/10.1007/s00467-019-04256-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-019-04256-0