
Abstract
Hemolytic uremic syndrome (HUS) is defined as a triad of noninmune microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury. The most frequent presentation is secondary to Shiga toxin (Stx)-producing Escherichia coli (STEC) infections, which is termed postdiarrheal, epidemiologic or Stx-HUS, considering that Stx is the necessary etiological factor. After ingestion, STEC colonize the intestine and produce Stx, which translocates across the intestinal epithelium. Once Stx enters the bloodstream, it interacts with renal endothelial and epithelial cells, and leukocytes. This review summarizes the current evidence about the involvement of inflammatory components as central pathogenic factors that could determine outcome of STEC infections. Intestinal inflammation may favor epithelial leakage and subsequent passage of Stx to the systemic circulation. Vascular damage triggered by Stx promotes not only release of thrombin and increased fibrin concentration but also production of cytokines and chemokines by endothelial cells. Recent evidence from animal models and patients strongly indicate that several immune cells types may participate in HUS physiopathology: neutrophils, through release of proteases and reactive oxygen species (ROS); monocytes/macrophages through secretion of cytokines and chemokines. In addition, high levels of Bb factor and soluble C5b-9 (sC5b-9) in plasma as well as complement factors adhered to platelet-leukocyte complexes, microparticles and microvesicles, suggest activation of the alternative pathway of complement. Thus, acute immune response secondary to STEC infection, the Stx stimulatory effect on different immune cells, and inflammatory stimulus secondary to endothelial damage all together converge to define a strong inflammatory status that worsens Stx toxicity and disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hemolytic uremic syndrome (HUS) is defined as a triad of noninmune microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury. The most frequent presentation is secondary to Shiga toxin (Stx)-producing Escherichia coli (STEC) infection, which has been termed postdiarrheal, epidemiologic, or Stx-HUS. Several years of investigation have established that it is a distinct clinical entity, in which Stx is the necessary etiological factor. Besides, STEC infections can lead to different outcomes, i.e., from self-limited gastrointestinal infection with no systemic complications to incomplete or complete HUS forms, or even a very aggressive presentation with neurologic involvement, which accounts for the 1–4% of mortality during the acute phase. Thus, several researchers during recent decades have demonstrated that host factors, particularly the inflammatory response, contribute to the pathogenesis of the disease. The aim of this work is to review the current evidence about the involvement of inflammatory components as central pathogenic factors that could determine the outcome of STEC infections. The understanding regarding how a specific mediator or cellular mechanism contributes to HUS development is the result of various experimental approaches, that as pieces of a puzzle include: the increasing knowledge about the mechanisms of Stx injury at cellular and subcellular levels obtained from in vitro studies; observations in animal models of STEC infections or Stx administration, even when none of them reproduce all the pathogenic components of HUS; and studies on Stx-HUS patients, which are often limited by the small size of the patient cohorts and/or the lack of serial sampling, but mainly because clinical studies do not allow distinction between causal or associative relationships of inflammatory abnormalities and disease.
Mechanism of Shiga toxin (Stx) action
Stx is formed of heterodimers consisting of one enzymatically active A subunit and a complex of five B subunits. The A subunit is a single-site RNA N-glycosidase for the 28S rRNA of the mammalian ribosome. Toxin binding to cells is mediated by B subunits, which primarily associate with the membrane neutral glycosphingolipids- globotriaosylceramide (Galα1-4Galβ1-4Glcβ1-1Cer (Gb3)) [1] and globotetraosylceramide (GalNAcβ1-3Galα1-4Galβ1-4Glcβ1-1Cer (Gb4)) [2]. These glycosphingolipids, which are considered the Stx-specific receptors, are located primarily in segments of the plasma membrane named lipid rafts that are rich in cholesterol, lipid-modified proteins, and transmembrane proteins. Gb3 is heterogeneous and displays variability in the fatty acid chain length and the degree of bond saturation and hydroxylation. Therefore, cells have different susceptibility to the toxin not only due to the presence or absence of Gb3 but also to the nature of the surrounding environment of the Gb3 receptor in the cell membrane and structural differences in the toxin receptor. For example, it has been suggested that the presence of cholesterol in the lipid rafts increases toxin binding, and expression of Gb3 isoforms with long-chain unsaturated fatty acids is associated with increased toxin sensitivity [3]. The particular composition of the Gb3 as well as of the cell membrane may define the strength of lateral interaction between them and the fate towards retrograde translocation of the toxin (sensitive cells) or towards its transportation to the lysosomes (not-sensitive cells) [4]. The binding step is followed by the retrograde transport of the A subunit to the endoplasmic reticulum (ER) and the Golgi apparatus, and the interruption of mRNA translation in the ribosome [5]. Thus, Stx has a direct effect on susceptible tissues by promoting cell injury, mainly due to inhibition of protein synthesis. Besides, Stx can induce a broad inflammatory response at lower concentrations than those needed to inhibit protein synthesis [6]. In fact, the activation of these ribotoxic and ER stress responses may produce alterations in the host signal transduction, thus affecting the expression of certain primary response genes (such as cytokines), or may initiate apoptotic signaling pathways, in part via the mitogen/stress-activated protein kinase pathway(s) [7].
In this sense, it has been shown that Stx treatment of a renal epithelial cell line (VERO cells) or undifferentiated monocytic THP-1 cells leads to the inhibition of protein synthesis. However, VERO cells are much more sensitive than THP-1 cells, and are used as one of the most sensitive ways to detect and quantify Shiga toxins. On the other hand, when THP-1 are differentiated into macrophages an equivalent dose of Stx triggers a transient increase in total protein synthesis, particularly TNF-α expression [8]. In addition, human intestinal epithelial cells treated with a dose of Stx that mediates only 10% cell death secrete the chemokine IL-8 [8]. Thus, Stx can be present at concentrations that do not affect cell viability, but still can trigger cytokine and chemokine induction within specific tissues during HUS.
In conclusion, Stx affects Gb3-positive cells mainly through specific damage to ribosomal 28S RNA. This effect is sufficient to activate stress kinase cascades that trigger cytokine/chemokine expression, and/or to induce cytotoxicity by inhibition of protein synthesis.
STX in vitro effects on different target cells
Endothelial and renal cells
Because of the pivotal role of endothelial and renal injury in Stx-HUS pathogenicity, the reciprocal influence between the cytotoxic direct effect of Stx and the inflammatory response in these tissues has been extensively studied.
In fact, the vascular damage triggered by Stx not only promotes the release of thrombin and increases fibrin concentrations but also induces the production of cytokines and chemokines by endothelial cells. High levels of plasminogen activator inhibitor-1 (PAI-1) block fibrinolysis and accelerate thrombosis [9]. Occluded vessels result in increased shear stress forces that inhibit the processing of von Willebrand factor (vWF) multimers by the metalloprotease ADAMTS13 [10], activate platelets, and augment thrombus formation [9]. In addition, Stx not only increases protein secretion of the vasoconstrictive peptide endothelin-1 (ET-1) [11], the endothelial adhesion molecule E-selectin, intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) [12, 13], but also induces the secretion of chemokines, such as IL-8, chemokine ligand (CCL) 2/monocyte chemotactic protein 1 (CCL2 or MCP-1) and human platelet factor-4 (PF4)/ chemokine (C-X-C motif) ligand 4(PF-4 or CXCL4), which increase leukocyte migration and adhesion.
Importantly, Stx-mediated cytokine regulation increases Gb3 expression on target cells, thus affecting their sensitivity to the toxin [14, 15].
In normal conditions, the endothelium exhibits a vasodilatory profile, is thromboresistant, anti-adhesive, and anti-inflammatory. In contrast, the incubation of endothelial cells with Stx induces secretion of IL-8 and MCP-1, both of which contribute to leukocyte adherence, and apoptosis [16]. These alterations lead to a leaky glomerular endothelium that could be responsible for proteinuria and leakage of cytokines, chemokines, and other biological markers of disease into the urine [17].
Because the kidney is one of the main target organs, Stx toxicity on different cellular types has been studied. It has been demonstrated that glomerular endothelium, proximal tubular epithelial and mesangial cells, and podocytes express Gb3 on their cell surfaces; therefore they are susceptible to toxin-mediated injury [18].
Several authors have shown that the epithelial cells of proximal tubules are sensitive to picomolar concentrations of Stx, which triggers apoptosis, inhibits water absorption and induces secretion of inflammatory cytokines, such as TNF-α, IL-1, and IL-6 in vitro. By contrast, in mesangial cells from human glomeruli, Stx inhibits protein synthesis and proliferation without altering cell viability [19].
In addition, podocytes are specialized epithelial cells that participate in the renal filtration barrier. They have extended foot processes, which wrap around the glomerular capillaries to form filtration slits, and are also separated from the glomerular endothelium by the basement membrane and glycocalyx [19]. Podocytes express Gb3 and are sensitive to picomolar concentrations of Stx. Low concentrations of Stx enhance expression of ET-1 in cultured podocytes, altering the hemodynamics [20]. Also, however, it has been reported that Stx decreases the secretion of vascular endothelial growth factor (VEGF), a potent angiogenic factor, and this could affect glomerular filtration, contributing to proteinuria [19, 21, 22].
In this way, injured epithelial and endothelial renal cells contribute to an inflammatory microenvironment that sensitizes glomerular endothelial cells to the cytotoxic effects of Stx, leading to acute kidney damage.
Leukocytes
Blood polymorphonuclear neutrophils (PMN) and monocytes/macrophages are sensitive to Stx and may be key players in HUS outcome.
Polymorphonuclear neutrophils (PMN)
The capacity of PMN to bind Stx is controversial. Even though several reports suggest an absence of Stx-binding to PMN, there are many results that demonstrate direct effects of Stx on PMN from healthy donors. Among these, it was shown that Stx delays apoptosis, induces reactive oxygen species (ROS) production, increases expression of activation markers such as CD11b and CD66b and triggers the formation of extracellular traps (NETs) [23]. Other authors have demonstrated that PMN show an oxidative response and degranulation upon incubation with Stx, followed by a subsequent hyporesponsiveness to a second activating stimulus interleukin (IL)-8 or phorbolmyristate acetate (PMA). Moreover, PMN treated with Stx release cytokines and chemokines such as IL-8, macrophage inflammatory protein1-betha (MIP-1β), tumoral necrosis factor-alpha (TNF-α), and granulocyte-colony stimulating factor (G-CSF) [24].
Monocytes/macrophages
Human monocytes/macrophages are less sensitive than epithelial and endothelial cells to Stx, which could be related to the failure of Gb3 to associate with lipid rafts and to direct Stx to the ER [5]. Alternatively, Stx appears to be routed into a degradative pathway in both macrophages and dendritic cells of human origin [25]. Associated with this pathway, Stx induces the expression of cytokines, such as IL-1β, IL-6, IL-8, and TNF-α in human monocytes [8, 26] and murine peritoneal macrophages [27] in vitro. In contrast to primary cells, a human myeloid leukemia cell line, THP-1 cells, expresses abundant Gb3 on the cell membrane and is susceptible to apoptosis by Stx through the activation of caspases 2, 6, 8, and 9 [25]. When THP-1 cells are differentiated to macrophage-like cells, Stx is able to activate pro- and anti-apoptotic signaling cascades simultaneously [28].
Involvement of inflammatory response in Stx-HUS: contributions from animal models
Animal models of HUS are based on oral infection with STEC [29, 30] or intravenous/intraperitoneal injection with purified Stx with or without lipopolysaccharide (LPS), given that LPS is an outer membrane component of Gram-negative bacteria and a strong inflammation inducer [31]. These models usually exhibit one or more of the three hallmarks of HUS: thrombocytopenia, hemolytic anemia, and renal failure. While all mouse models show histopathological changes in kidneys, including acute tubular necrosis and matrix expansion, vascular congestion, and interstitial inflammation [32], some of them (i.e., Stx plus LPS) also reproduce other systemic alterations typically observed in HUS patients, such as platelet activation, fragmented erythrocytes, neutrophilia, neurological symptoms, and intestinal damage, but none reproduce all of them [33,34,35,36]. Because mouse models have been particularly valuable to study immune and inflammatory responses during human diseases, we will focus on reports in mouse models that shed light on the contribution of the inflammatory response to HUS pathogenesis [37] (Table 1).
Mouse models based on STEC infections
STEC adhere to the apical membrane of colonic epithelial cells and produce several pathogenic factors that, besides their specific function, trigger an acute inflammation in the intestine through different pathways [38]. These factors include LPS, the surface protein intimin, Stx, and H7 flagellin. Thanks to the availability of knock-out (KO) mice with breakdown in different components of the immune response, it has been possible to elucidate their role in HUS pathogenesis.
For example, engineered KO mice lacking a component of the signaling pathway shared by LPS and flagellin showed the highest severity of clinical signs after infection with STEC compared to controls or engineered KO mice lacking a component exclusive of the LPS signaling pathway [39]. In fact, the first mice have a defective bacterial clearance and a leakier intestinal barrier, thus allowing systemic spread of Stx and LPS [40, 41]. These results support the importance of the initial steps of intestinal inflammation in the resolution of STEC infection.
In conclusion, although excessive inflammation may be deleterious, an initial response to STEC in the gut could activate cytoprotective mechanisms necessary for bacterial clearance, such as PMN recruitment or production of IL-6 and chemokine ligand 1 /keratinocyte-derived chemokine (CXCL1/KC), the murine IL-8 mimic [42].
Another STEC pathogenic factor is the pore-forming cytolysin hemolysin (Hly). At the intestinal level, it has been demonstrated that Hly induces a biphasic response in residing mast cells, thus leading to the release of cytokines and inflammatory mediators, including leukotrienes (LTs) [43]. In line with this observation, it was shown that LTs concentration increases in the intestinal mucosa during STEC infection in mice and rabbits [44, 45]. Moreover, LTs pre-treatment of STEC-infected mice affects intestinal integrity and increases the susceptibility to HUS [45]. In addition, a more severe score of intestinal damage after intragastrical infection of mice with STEC was reported during experimental vitamin A deficiency [30]. Altogether, these results support the conclusion that the disruption of the mucosa may facilitate the passage of pathogenic factors, mainly Stx, to the bloodstream.
Thus, mouse models based on STEC infection support the idea that those factors that contribute to preserve mucosal integrity and trigger a controlled inflammatory response are not only beneficial to the host, but necessary to avoid systemic complications secondary to STEC infections.
Mouse models based on systemic inoculation of Stx
Mouse models have also been helpful in the study of the systemic action of purified Stx, as well as in separating the actions of Stx and LPS in HUS. Stx is produced in the intestine and translocated across the intestinal epithelium by a still not completely understood mechanism. Once Stx enters the bloodstream, it can interact with endothelial and epithelial cells of the kidney, leukocytes, and central nervous system (CNS) cells. The intravenous injection of Stx into mice has allowed demonstration that Stx induces an early and marked neutrophilia, which positively correlates with renal damage; and that this phenomenon is a consequence of several events, including acceleration in the release of bone marrow cells, increase in the proliferation of myeloid progenitor cells, and prevention of migration into tissues [46]. Besides, peripheral PMN show functional and phenotypic parameters of activation [46, 47]. The relevance of PMN activation in HUS is indirectly supported by experiments done in mice treated with retinoic acid, the precursor of vitamin A. These mice exhibit an increased absolute number of circulating PMN with an increased capacity to produce ROS, and are more sensitive to Stx. On the other hand, vitamin A-deficient mice, which exhibit a reduced number of PMN and a decreased capacity to produce ROS, have a better outcome after STEC infection than control mice [30]. Taken together, these results highlight the direct relationship between functionality of PMN and susceptibility to Stx toxicity. In line with this, it has been demonstrated that endogenous or exogenous glucocorticoids can attenuate Stx2 toxicity and HUS severity in mice, at least in part, by restraining PMN activation [48]. Besides, mice depleted of PMN present a reduced sensitivity to Stx-dependent renal toxicity and lethal effects [37]. All of these results indicate that neutrophilia is not merely an epiphenomenon, but contributes to the pathophysiology of Stx by exacerbating Stx-induced renal damage and mortality [37].
Stx injection into mice induces a strong oxidative stress both systemically and locally at the kidney level [49]. ROS are prothrombotic, induce endothelial dysfunction and damage by lipid peroxidation, and the superoxide anion reduces the threshold for platelet activation to several stimuli [50]. PMN represent the main source of oxidative stress during HUS and antioxidant treatment improves platelet response, renal damage, and survival after Stx injection [49]. The above evidence supports the hypothesis that an imbalance of antioxidants and ROS contributes to endothelial damage and renal failure [51], both of them central pathogenic events during Stx-HUS.
In conclusion, these experimental approaches have been important in demonstrating that Stx-dependent injury is enough to trigger a strong inflammatory response in which circulating PMN are activated early and intensely. Considering that the direct effects of Stx on PMN are light or mild, it is reasonable to hypothesize that the effect of PMN activation by Stx in vivo is the result of direct but also indirect effects, mainly secondary to Stx-mediated endothelium damage.
Furthermore, modeling HUS by intravenous injection of Stx has also allowed it to be demonstrated that monocytes play a pathogenic role [52]. In fact, this population would participate through secretion of IL-1 and TNF-α, which in turn sensitize the endothelium to the cytotoxic effect of Stx, but also as local players of inflammation in the glomeruli. In this regard, chemokine receptor-1 (CCR1) KO mice have showed an increased survival rate after Stx injection compared with CCR5 KO and control mice [53]. Both receptors share their main ligands (chemokine ligand (CCL) 3 / macrophage inflammatory protein 1α (MIP-1α), and CCL5 / regulated on activation, normal T cell expressed and secreted (RANTES)), but while CCR1 is expressed on monocytes, macrophages, and PMN, CCR5 expression is absent in PMN. Analysis of tissue-associated leukocytes has demonstrated that CCR1 KO mice have a delayed and lesser increase of circulating leukocytes, and a delayed peak of plasmatic TNF-α and IL-6 compared to control mice [53]. All these results suggest that CCR1 may be necessary for cell recruitment and amplification of local and systemic inflammatory responses in HUS [53]. Therefore, locally secreted chemokines participate in the accumulation of inflammatory cells at the kidney level, and amplify the inflammatory processes instrumental to the activation of renal microvascular endothelial cells.
In the mouse model of HUS produced by injection of Stx and LPS, the analysis of gene activation on endothelial and renal tubule cells has showed that, whereas Stx enhances the activity of LPS, LPS is the primary inducer of cytokines and chemokines, such as CCL2/monocyte chemotactic protein 1 (MCP-1), CCL3/MIP-1α, CCL5/RANTES [54]. Similarly, the PMN chemotactic factors chemokine (C-X-C motif) ligand (CXCL) 1/KC and CXCL2/MIP-2 are induced at the transcriptional level in the kidneys by LPS and enhanced by Stx [55]. All these chemokines lead monocytes to the extraglomerular space, while PMN also migrate into the glomeruli and set the stage for a local broad inflammatory response. In line with this interpretation, the simultaneous neutralization of these chemokines inhibits LPS/Stx-induced monocyte accumulation and fibrin deposition in the kidneys [54]. LPS is also the initial primary elicitor of renal coagulation and thrombosis, but Stx enhances these effects and is the lethal agent of STEC [56]. Considering that the mouse model by Stx/LPS inoculation better resembles HUS in humans (including the glomerular involvement and thrombotic response), it prompts the question as to whether traces of LPS, which is one of the biological agents with highest inflammatory activity, could gain access to the bloodstream even when STEC infection does not cause bacteremia.
In contrast to the clear participation of inflammation during systemic and endothelial complications after STEC infections, the pathogenic mechanisms driving towards CNS alterations are controversial or poorly understood. In this regard, the injection of purified Stx directly into CNS parenchyma induces similar symptoms to those observed in STEC-inoculated mice [34] and rabbits [22] as well as in Stx-injected rabbits [35], such as lethargy, hind-leg weakness, or paralysis. In addition, ultrastructural studies of the brains of Stx-treated mice and rats exhibit neuronal damage, including demyelinated axons, cytoplasmic edema, and degenerative phenotypes [34, 36]. These observations suggest that pro-inflammatory responses can alter the blood–brain barrier (BBB) whereby Stx could gain access to CNS parenchyma where it asserts its toxicity. In fact, BBB weakening was evidenced by the reduction of AQP4 in rats [57] and mice [58], and has been associated with the increase of serum TNF-α in STEC-inoculated mice [59], rabbits [22], and Stx2-injected rabbits [35].
In contrast, other studies do not support a direct effect of Stx on brain endothelial cells [60] and suggest that energy depletion by lack of glucose and oxygen intake or electrolyte disorders and/or inflammatory mediators could damage the brain [60]. Furthermore, symmetrical microglial activation occurring in many parts of the brain suggests that local ischemic or hemorrhagic events are not responsible for the neurological damage, but operation of a more global process. In this regard, even though mouse astrocytes lack Gb3 expression [34, 36], a severe swelling and cellular breakdown was observed following Stx injection, thus suggesting that these cells are indirectly damaged. Moreover, selective deletion of Gb3 expression in renal tubule cells resulted in around 65% of mice death due to neurological complications [61]. These results suggest that neurological damage is not just a secondary response to kidney damage, and that Gb3-expressing cells that do not reside in the kidney or endothelium play a significant role in Stx-mediated complications and death [61].
In summary, the major advantage of animal models is the possibility of performing cause–effect associations. In addition, a particular contribution of HUS mouse models developed by STEC infection or Stx/Stx + LPS intravenous injection is that they allow analysis of the early course of disease, which normally takes place before clinical studies, since patients are studied at the moment of HUS diagnosis. This may also be the cause of some differences found in both cases (animals vs. humans) (Table 1).
Evidence of inflammatory response in Stx-HUS patients
In the following paragraphs, the involvement of different components of the inflammatory response in patients with Stx-HUS is summarized (Table 1). We have highlighted those studies that provide the strongest support for a pathogenic role.
Leukocytes
Polymorphonuclear neutrophils (PMN)
An increased PMN count on peripheral blood at diagnosis is one of the foremost and most consistent parameters of inflammation associated with a poor prognosis [62, 63], and renal biopsies carried out during the early phase of the disease commonly show infiltration of PMN in Stx-HUS patients [64, 65]. In this regard, different pathogenic roles have been proposed for PMN. Among them, PMN could participate through the delivery of Stx to the glomerular microcirculation [66], a process driven in part by an increased expression of Toll-like receptors on circulating PMN [67]. However, PMN may also directly contribute to renal inflammation and endothelial injury during HUS, based on their great cytotoxic potential through exocytosis of granules-containing proteases and other enzymes, as well as high release of ROS upon activation.
The activation of PMN results in mobilization of their granules and a rapid upregulation on membrane expression of the molecules contained in them, which can be analyzed by flow cytometry. Mouse models of HUS have been particularly useful to demonstrate PMN activation during very early phases of Stx intoxication, as was previously described [46]. In contrast, peripheral PMN from patients at HUS diagnosis show decreased expression of membrane antigens, together with the presence of PMN-derived proteases in the serum, according to a temporal biphasic process. In this regard, data reporting decreased intracellular content of enzymes and antigens supports the concept that PMN from Stx-HUS patients have been previously activated and degranulated [68, 69]. In line with these results, it has been proposed that a strong initial activating stimulus would induce a strong activation of PMN, that in turn would drive to a more severe clinical course. Consequently, PMN deactivation at admission shows a close correlation with the severity of renal dysfunction achieved during the acute period [63].
Another potentially pathogenic mechanism is the release of NETs in a ROS/NADPH-dependent manner by PMN. NETs are a meshwork of DNA fibers comprising histones and granule proteins, such as elastase, myeloperoxidase (MPO), pentraxin, and lactoferrin, each of them with strong antimicrobial and/or immunomodulatory properties [70]. Even though NETs participate in the control of bacteria dissemination, high amounts of them seem to be associated with pathophysiological conditions, thus suggesting that NETs contribute to collateral damage within inflamed tissues [71]. When these structures are formed inside the microvasculature, they act as a stimulus for thrombus formation [72]. Besides, NETs induce adhesion, activation and aggregation of platelets that finally promote fibrin deposition, thus suggesting that NETs are a link between inflammation and thrombosis [71]. Interestingly, increased plasma levels of circulating free-DNA (cf-DNA) that co-localize with MPO are increased in plasma from Stx-HUS children [23]. In addition, increased levels of cf-DNA have also been reported in thrombotic microangiopathy (TMA) patients [73]. Moreover, a recent report showed that only those patients with Stx-HUS from a cohort of TMA have a decreased ability to degrade NETs secondary to a decreased nuclease activity, and decreased NET degradation is associated with disease activity in the TMA cohort [74]. Thus, a conserved or increased NET release by PMN in combination with attenuated degradation may adversely contribute to the kidney damage in Stx-HUS patients. In fact, it has been suggested that NETs could serve as scaffolding that trap platelets and express the prothrombotic tissue factor that initiates coagulation [75]. Moreover, several authors have reported that NETs can activate complement [76, 77] and this could enhance the Stx-induced complement activation, thus leading to C3b deposition onto glomeruli and platelets [78, 79], with the subsequent induction of microvascular thrombosis and kidney injury [80]. These adverse procoagulant, proinflammatory and complement-activating effects of NETs may be particularly severe in kidneys and fit well within the known pathology of Stx-HUS.
Oxidative mediators
PMN represent the main source of oxidative stress, which is removed or balanced by endogenous antioxidant compounds [81]. On the other hand, glutathione (GSH) is the most important intracellular antioxidant that acts as a reducing agent and protects cells by sequestering ROS and detoxifying the intracellular medium [82]. Therefore, the increase in oxidative stress parallels the decrease in the antioxidant capacity of the cell due to the loss of GSH [82].
Stx-HUS patients during the acute phase have higher levels of lipid peroxidation of red blood cells [83], higher amounts of proteins with signs of advanced oxidation [84], and higher concentrations of oxidized glutathione (GSSG) than healthy controls [85]. In parallel, a significant decrease in superoxide dismutase (SOD) activity is found in erythrocytes from HUS patients, and the addition of their own plasma to in vitro cultures further decrease SOD activity [86, 87]. In line with these results, Powell et al. studied the effects of vitamin E in a pilot study conducted in a small group of Stx-HUS patients. They observed that patients treated with vitamin E fared considerably better than those not treated. In view of the absence of side effects, they suggest further experience with this treatment to be conclusive [88]. Taken together, these studies show that acute renal failure can be triggered by ROS, but also that once established it may contribute to an oxidative imbalance.
Monocytes/macrophages
Stx-HUS patients also show a correlation between monocytosis and HUS severity [89], together with changes in the expression of chemokine receptors on these cells [89,90,91]. In particular, a selective depletion of circulating mononuclear leukocytes expressing the receptor for fractalkine/CX3CL1 (CX3CR1) was observed, that correlates with the severity of renal failure [92]. Moreover, CX3CR1-positive leukocytes were observed in renal biopsies from Stx-HUS patients. Altogether these results suggest that the interaction of CX3CR1-positive cells with CX3CL1 present on activated kidney endothelial cells may contribute to renal injury in HUS [92]. In addition, monocytes isolated from Stx-HUS patients show a differential pattern of cytokine [90] and chemokine [62, 93] production in vitro. In fact, production of TNF-α and IL-10 by monocytes increases in parallel with the severity of disease in Stx-HUS children, in such a way that patients with moderate-to-severe disease have the greatest number of TNF-α-producing monocytes [90].
All these data demonstrate that monocytes also suffer activation during Stx-HUS. Because histological studies of biopsy specimens from Stx-HUS patients show the presence of monocytes in glomeruli [65], their involvement in glomerular endothelial cell damage has been proposed.
Soluble mediators
Soluble mediators might play an important role in the pathogenesis of Stx-HUS and may be also useful to predict the severity of HUS. Their search has been an intensive field of research because the collection and preservation of the samples is simple and allows new parameters to be evaluated a posteriori. However, these reports do not always allow a causal or associative relationship between inflammatory abnormalities and the disease to be distinguished, and it is difficult to unify results between the different studies since the cytokines are temporarily secreted and have a fine cross-regulation system. Consequently, the concentration of cytokines found is highly dependent on the time of sampling in relation to the onset of disease and also highly variable among the individuals.
Chemokines
It has been reported that the influx of monocytes and PMN into glomeruli may be an important event in the initiation, prolongation, and progression of glomerular endothelial cell damage in Stx-HUS patients. In this regard, some clinical studies have found elevated levels of specific chemokines in urine samples from Stx-HUS patients, such as MCP-1 and IL-8 [65]. In addition, plasma levels of G-CSF and the chemokines epithelial cell-derived neutrophil-activating protein-78 (ENA-78), growth-related oncogen-alpha (GRO-α), MIP-1β and MCP-1, are increased in pediatric patients with STEC infections [93]. However, only those children who progress to HUS present abnormally increased circulating levels of G-CSF and SDF-1 [9], and decreased ENA-78 concentrations.
Cytokines
Several studies have consistently reported that Stx-HUS patients have higher plasma levels of TNF-α, IL-6, IL-8, and G-CSF compared to healthy controls [94, 95]. Moreover, circulating levels of TNF-α and IL-6 correlate with the severity of Stx-HUS and the occurrence of extrarenal complications. Patients who progress to severe renal dysfunction have a 10-fold increase in IL-6 compared to those who maintain diuresis [94]. Besides, elevated concentrations of IL-6 and soluble TNF-receptor I (sTNFRI) are associated with the occurrence of encephalopathy [96].
On the other hand, while some authors have found lower levels of IL-10 in STEC-infected patients, others have found that most patients with typical signs of HUS have elevated levels of circulating IL-10 compared to those without HUS [97]. This discrepancy could represent differences in the timing of serum sample collection with respect to diarrhea-HUS onset among different studies, but also it could be speculated that increased serum IL-10 might lead to a prolongation of the intestinal infection, which in turn could result in a higher possibility of HUS outcome.
It has been reported that Stx-HUS children have reduced levels of angiopoietin-1 (anti-inflammatory) and increased levels of angiopoietin-2 (inflammatory) during the prodromal phase, that worsen with the progression of microangiopathy [98]. Alongside this, serum concentrations of endothelin and thrombomodulin, which are molecular markers of endothelial damage, are also elevated in Stx-HUS patients preceding the deterioration of renal function [99, 100]. Increased levels of thrombomodulin correlate with serum levels of IL-6, IL-8, IL-10 and endothelin. Endothelin can induce microthrombosis by the increased synthesis of vWF, has vasoconstrictor properties and is also able to increase the synthesis of IL-8. These findings support the concept that subclinical endothelial dysfunction precedes HUS onset.
These findings certainly indicate that toxin-mediated endothelial injury results in a prothrombogenic intravascular environment and these alterations may potentiate systemic inflammation.
In this regard, Shimizu et al. identified five serum biomarkers, namely insulin growth factor-binding protein-2, angiopoietin-2, soluble IL-6 receptor, sTNFR type II (sTNFRII), and matrix metalloprotease protein-3, whose levels increase with HUS outcome and correlate with severity [95]. In addition, it has been reported that increased plasma concentrations of procalcitonin [101] are associated with severity of renal dysfunction during HUS.
Regarding T cell-specific immune response, no imbalance of Th1 vs Th2 cytokines is observed in patients, given that serum levels of IL-2, IL-4, IL-13 and IFN-gamma are low and comparable between Stx-HUS and control groups. However, TGF-β concentration is higher in STEC-infected children than HUS children [94].
Although further studies to determine the role of all these soluble factors on the pathogenesis of Stx-associated TMA are needed, it is clear that a strong inflammatory response is ongoing during HUS development.
Complement
Although the classical lectin and alternative pathways of complement are activated by various stimuli during disease, all of these converge with the generation of the membrane terminal attack complex (MAC). MAC can exist as a membrane-bound form on cells and tissues, or as a soluble C5b-9 complex (sC5b-9) in the fluid phase.
While initial studies of Stx-HUS patients showed normal levels of C3 and C4 factors, others reported low levels of C3, at least in a subset of patients, and/or increased levels of complement degradation products (C3b, C3c, and C3d) [102]. More recently, clinical studies in Stx-HUS have shown activation of the alternative pathway based on high plasma levels of Bb factor and sC5b-9, which normalize after recovery [103, 104].
In addition, Karpman’s group found complement activation on platelet–leukocyte complexes and platelet- and monocyte-derived microparticles in the circulation of Stx-HUS children [79], as well as complement-coated red blood cell-derived microvesicles [105].
The activation of the complement system is probably a consequence of the acute phase reaction to the STEC infection [106], but also the Stx-mediated direct inhibition of complement regulator Factor H, thus leading to dysregulation of the alternative complement pathway [78, 104, 107]. On the other hand, endothelial cells that are exposed to Stx in vitro increase expression of P-selectin, which binds and activates C3 via the alternative pathway [78]. The C3a that is produced following the cleavage of C3 exacerbates the activation of the alternative pathway, reduces the expression of thrombomodulin and promotes thrombus formation. These findings indicate that the complement system contributes to the abnormal vascular function during Stx-HUS.
Although the pathogenic role of complement activation is still poorly understood, new data from the most recent outbreaks have suggested a relationship between complement activation and CNS compromise. In fact, HUS systemic presentation varies greatly among patients, but Stx-HUS-related early death is generally related to CNS compromise, which is as high as 20% in pediatric Stx-HUS [108, 109], and even affected 50% of adults during the German O104 outbreak [110].
During the large German outbreak in 2011, the C5 complement inhibitor, eculizumab, which is licensed for paroxysmal nocturnal hemoglobinuria and atypical HUS, was used mainly in severe Stx-HUS or patients with neurological compromise. Although two large series showed similar outcomes in patients treated or not with eculizumab [111], subsequent smaller and uncontrolled series have reported a rapid improvement with eculizumab [112,113,114]. Interestingly, cumulative experience in Stx-HUS has shown that the early use of eculizumab can improve the clinical course of neurological symptoms but not other parameters, such as the length of dialysis or duration of thrombocytopenia [114, 115].
Although complement function (CH50 and APH50) is abolished following eculizumab treatment, sC5b-9 decay is not affected, probably due to its half-life and the incomplete inhibition of the production of complement protein by renal cells [106]. In addition, eculizumab only blocks the terminal sequence of the complement system but does not interfere with C3 activation, which can also lead to cell damage via opsonization and subsequent activation of inflammatory cells. A reduction of C3 activation (C3d) indicates that the initial triggers for complement activation (tissue damage, Stx-induced factor H inhibition) are only transiently active.
The question of whether Stx-HUS children really benefit from eculizumab treatment can only be answered by future randomized trials with larger numbers of well-defined groups of patients comparing early eculizumab treatment with standard care.
There is little evidence of cellular death in the brain, and permanent neurologic damage is typically not observed in human patients after resolution of the acute symptoms. Altogether these results indicate that acute proinflammatory reaction might be closely associated with the pathogenesis of brain injury in Stx-HUS. Following this reasoning, and because corticosteroids are potent anti-inflammatory agents, some therapeutic protocols have included methylprednisolone (mPSL) treatment of patients with STEC encephalopathy [116, 117]. In fact, mPSL pulse therapy increased the probability of a good outcome in an Argentinean group of patients [117]. Based on similar results reported by Takanashi et al. during the 2011 outbreak in Japan, the Japanese Society of Pediatric Nephrology have recommended that mPSL pulse therapy could be considered in patients with severe STEC encephalopathy. Although the efficacy of this treatment has not been completely established, severe encephalopathy is a predictor of poor outcome with regard to neurologic function and/or survival [118].
Some evidence suggests that neurological complications could be more related to systemic and local inflammatory reactions than to thrombosis, ischemic changes or direct toxic effect of Stx on neurons. In this regard, the postmortem neuropathological investigation of brains from five patients who died during the German outbreak in 2011 showed a slightly increased activation of microglia and a higher neuronal expression of IL-1β and Gb3 [119]. In the same line of reasoning, elevated serum levels of tau protein are seen in patients with STEC encephalopathy compared with STEC O111/HUS patients without encephalopathy, patients with non-STEC-related acute encephalopathy and healthy controls [120]. Tau is a microtubule stabilizing protein primarily localized in CNS neurons, but it is also expressed at low levels in astrocytes and oligodendrocytes. Because deterioration of the BBB occurs in the early stage of HUS, tau protein released from injured axons might leak rapidly into the vascular space, thus resulting in a serum-dominant increase in tau protein. The same authors reported that serum tau protein levels are positively correlated with proinflammatory cytokines, including neopterin, IL-6, sTNFRI, and sTNFRII [120].
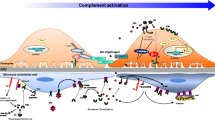
On the other hand and based on experimental studies, it has been proposed that neurological compromise represents a combined effect of Stx-induced vascular injury, endothelial dysfunction, hypertension and electrolyte disorders. Overall, these findings support the idea that the clinical outcome is caused not only by the direct effects of Stx, but also by secondary effects induced by the inflammatory response and the complement system (Fig. 1).

Involvement of inflammatory response in the pathogenic mechanism of Stx-HUS. As a consequence of Stx-direct endothelial damage and Stx/STEC-triggering of immune response, inflammatory and thrombotic responses are mutually stimulated. a Stx binds to monocytes and endothelial cells (EC) promoting their activation, maturation and secretion of cytokines and chemokines. b Cytokines released cause up-regulation of adhesion molecules and Gb3 receptor in EC. c Activation of EC leads to secretion of thrombotic factors that induce platelet aggregation and degranulation. d Stx internalization in activated EC induces inhibition of protein synthesis and cellular death. e Several factors released by activated endothelium, monocytes and platelets collaborate in PMN activation. f Activated PMN release their granule content, produce ROS and NETs, adhere to EC and, together with platelets and monocytes, potentiate Stx-induced EC damage. ULvWF: ultra-large von Willebrand factor, TF: tissue factor, TNF-α: tumor necrosis factor-α, IL-1β: interleukin-1 β, PF-4:platelet factor 4, MIP-α macrophage inflammatory protein-α, MCP-3:monocyte chemoattractant protein-3, IL-8: interleukin-8, ICAM-1: intercellular adhesion molecule-1. Adapted from [121]
Conclusions
Children infected with Stx-producing bacteria can develop a clinical pathological entity named Stx-HUS. After the binding of Stx to Gb3 receptors, the A subunit undergoes retrograde transport and interacts with ribosomes leading to the inhibition of protein synthesis and causing injury, apoptosis, death, and /or activation of the susceptible cells. The central pathogenic feature of Stx-HUS is glomerular endothelial damage and the development of microangiopathic thrombosis caused directly by Stx. In addition, during the last decade several studies in animal models have showed the simultaneous activation of a strong inflammatory response causing the release of cytokines and chemokines, the recruitment of leukocytes in the kidney and the activation of complement and thrombotic cascades during Stx-HUS outcome. In parallel, several studies in Stx-HUS patients have found immunological parameters indicative of an inflammatory status. Importantly, studies in animals have demonstrated that the inflammatory response is able to modulate Stx-direct cytotoxic effects, and that PMN and monocytes play a central pathogenic role through their high pro-oxidative and cytotoxic potential. In the same line, research studies in Stx-HUS patients have showed a strong correlation between alterations of those immunological parameters during the acute phase and the clinical outcome of the disease, thus reinforcing the concept that inflammation is another pathogenic mechanism and not merely an epiphenomenon. The analysis of those immune mechanisms using experimental models allows cause–effect relationships to be proven, which constitute the rational basis for development of future therapies. On the other hand, some areas are still poorly understood and need further research. The regulation of Stx production in the human gut, toxin transport from the intestine to target cells in the kidney and the pathogenic mechanism underlying CNS complications are some of the issues that require a better understanding for the development of early intervention strategies against HUS.
Key summary points
-
1.
Upon interaction of Stx with its specific receptor (Gb3) on target cells, mainly endothelial and epithelial renal cells, Stx can inhibit protein synthesis but also can trigger ribotoxic stress. In this way, Stx can induce cellular death but also synthesis of inflammatory mediators depending on cell type and toxin concentration.
-
2.
Animal models allow the demonstration that inflammatory response, mainly activation of PMN and monocytes/macrophages are central pathogenic factors and not merely an epiphenomenon.
-
3.
Studies on Stx-HUS patients show evidence of a strong inflammatory response that may accompany any other infectious disease. However, the special feature of this disease is the simultaneity with the Stx-direct endothelial damage. Stx-endothelial injury leads to platelet aggregation, thrombotic occlusion, and microangiopathic hemolytic anemia (the pathognomonic TMA lesion), which certainly potentiates inflammation, and this inflammation could contribute to endothelial damage, in a reciprocal positive and pathogenic feedback loop.
Multiple-choice questions (answers are provided following the reference list)
Choose the correct answer for each question:
-
1.
What are the consequences of Stx-Gb3 interactions on susceptible cells?
-
a)
This interaction leads to cell death or production of chemokines/cytokines depending on the target cell and toxin concentration.
-
b)
This interaction leads only to the production of chemokines/ cytokines in target cells.
-
c)
This interaction leads to death of target cells
-
d)
This interaction leads to apoptosis or proliferation of renal cells depending on the toxin concentration.
-
2.
How does STEC trigger the intestinal immune response?
-
a)
Although LPS, Hly, flagellin and intimin trigger an intestinal inflammatory response, Stx inhibits it.
-
b)
STEC triggers the intestinal immune response through Stx-production.
-
c)
STEC triggers the intestinal immune response through several bacterial components, mainly LPS, Hly flagellin, intimin, and Stx.
-
d)
STEC trigger the intestinal immune response through several bacterial components, however, Stx is irrelevant because Gb3 is absent from human gut.
-
3.
How could systemic inflammation participate in Stx-HUS development?
-
a)
Systemic inflammation induces an anti-thrombotic response.
-
b)
Systemic inflammation neutralizes Stx.
-
c)
Systemic inflammation is irrelevant for HUS outcome.
-
d)
Systemic inflammation and thrombotic response are mutually exacerbated.
-
4.
What have mouse models based on intravenous Stx and LPS injection contributed to our understanding?
-
a)
These models better reproduce clinical aspects of Stx-HUS compared to intravenous Stx injection, thus supporting the concept that systemic inflammation (triggered by LPS) plays a central pathogenic role during HUS.
-
b)
These models better reproduces gastrointestinal features of children infected with STEC, as LPS is a major component of STEC.
-
c)
These models do not contribute any additional or supplemental information compared to mouse models based on intravenous Stx injection.
-
d)
These models better reproduce neurological alterations of children infected with STEC, because LPS is neurotropic.
-
5.
According to evidence recorded from Stx-HUS patients, which components of the immune response are activated?
-
a)
Cytokine production and activation of the classical pathway of complement.
-
b)
Activation of all components of inflammation: PMN, monocytes, complement, cytokines and chemokines.
-
c)
Th1-specific cellular response and chemokine release
-
d)
Activation of local immune response in the gut, without systemic response.
References
Jacewicz M, Clausen H, Nudelman E, Donohue-Rolfe A, Keusch GT (1986) Pathogenesis of shigella diarrhea. XI. Isolation of a shigella toxin-binding glycolipid from rabbit jejunum and HeLa cells and its identification as globotriaosylceramide. J Exp Med 163:1391–1404
Samuel JE, Perera LP, Ward S, O’Brien AD, Ginsburg V, Krivan HC (1990) Comparison of the glycolipid receptor specificities of Shiga-like toxin type II and Shiga-like toxin type II variants. Infect Immun 58:611–618
Muthing J, Schweppe CH, Karch H, Friedrich AW (2009) Shiga toxins, glycosphingolipid diversity, and endothelial cell injury. Thromb Haemost 101:252–264
Khan F, Proulx F, Lingwood CA (2009) Detergent-resistant globotriaosyl ceramide may define verotoxin/glomeruli-restricted hemolytic uremic syndrome pathology. Kidney Int 75:1209–1216
Tesh VL (2010) Induction of apoptosis by Shiga toxins. Future Microbiol 5:431–453
Trachtman H, Austin C, Lewinski M, Stahl RA (2012) Renal and neurological involvement in typical Shiga toxin-associated HUS. Nat Rev Nephrol 8:658–669
Thorpe CM, Hurley BP, Lincicome LL, Jacewicz MS, Keusch GT, Acheson DW (1999) Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infect Immun 67:5985–5993
Foster GH, Armstrong CS, Sakiri R, Tesh VL (2000) Shiga toxin-induced tumor necrosis factor alpha expression: requirement for toxin enzymatic activity and monocyte protein kinase C and protein tyrosine kinases. Infect Immun 68:5183–5189
Petruzziello-Pellegrini TN, Yuen DA, Page AV, Patel S, Soltyk AM, Matouk CC, Wong DK, Turgeon PJ, Fish JE, Ho JJ, Steer BM, Khajoee V, Tigdi J, Lee WL, Motto DG, Advani A, Gilbert RE, Karumanchi SA, Robinson LA, Tarr PI, Liles WC, Brunton JL, Marsden PA (2012) The CXCR4/CXCR7/SDF-1 pathway contributes to the pathogenesis of Shiga toxin-associated hemolytic uremic syndrome in humans and mice. J Clin Invest 122:759–776
Nolasco LH, Turner NA, Bernardo A, Tao Z, Cleary TG, Dong JF, Moake JL (2005) Hemolytic uremic syndrome-associated Shiga toxins promote endothelial-cell secretion and impair ADAMTS13 cleavage of unusually large von Willebrand factor multimers. Blood 106:4199–4209
Bitzan MM, Wang Y, Lin J, Marsden PA (1998) Verotoxin and ricin have novel effects on preproendothelin-1 expression but fail to modify nitric oxide synthase (ecNOS) expression and NO production in vascular endothelium. J Clin Invest 101:372–382
Morigi M, Zoja C, Figliuzzi M, Foppolo M, Micheletti G, Bontempelli M, Saronni M, Remuzzi G, Remuzzi A (1995) Fluid shear stress modulates surface expression of adhesion molecules by endothelial cells. Blood 85:1696–1703
Morigi M, Galbusera M, Binda E, Imberti B, Gastoldi S, Remuzzi A, Zoja C, Remuzzi G (2001) Verotoxin-1-induced up-regulation of adhesive molecules renders microvascular endothelial cells thrombogenic at high shear stress. Blood 98:1828–1835
Obrig TG, Louise CB, Lingwood CA, Boyd B, Barley-Maloney L, Daniel TO (1993) Endothelial heterogeneity in Shiga toxin receptors and responses. J Biol Chem 268:15484–15488
Kaye SA, Louise CB, Boyd B, Lingwood CA, Obrig TG (1993) Shiga toxin-associated hemolytic uremic syndrome:interleukin-1 beta enhancement of Shiga toxin cytotoxicity toward human vascular endothelial cells in vitro. Infect Immun 61:3886–3891
Zoja C, Angioletti S, Donadelli R, Zanchi C, Tomasoni S, Binda E, Imberti B, te Loo M, Monnens L, Remuzzi G, Morigi M (2002) Shiga toxin-2 triggers endothelial leukocyte adhesion and transmigration via NF-kappaB dependent up-regulation of IL-8 and MCP-1. Kidney Int 62:846–856
Obrig TG (2010) Escherichia coli Shiga toxin mechanisms of action in renal disease. Toxins (Basel) 2:2769–2794
Williams JM, Boyd B, Nutikka A, Lingwood CA, Barnett Foster DE, Milford DV, Taylor CM (1999) A comparison of the effects of verocytotoxin-1 on primary human renal cell cultures. Toxicol Lett 105:47–57
Obrig TG, Karpman D (2012) Shiga toxin pathogenesis: kidney complications and renal failure. Curr Top Microbiol Immunol 357:105–136
Morigi M, Buelli S, Zanchi C, Longaretti L, Macconi D, Benigni A, Moioli D, Remuzzi G, Zoja C (2006) Shigatoxin-induced endothelin-1 expression in cultured podocytes autocrinally mediates actin remodeling. Am J Pathol 169:1965–1975
Ohara S, Kawasaki Y, Abe Y, Watanabe M, Ono A, Suyama K, Hashimoto K, Honda T, Suzuki J, Hosoya M (2012) Role of vascular endothelial growth factor and angiopoietin 1 in renal injury in hemolytic uremic syndrome. Am J Nephrol 36:516–523
Kita E, Yunou Y, Kurioka T, Harada H, Yoshikawa S, Mikasa K, Higashi N (2000) Pathogenic mechanism of mouse brain damage caused by oral infection with Shiga toxin-producing Escherichia coli O157:H7. Infect Immun 68:1207–1214
Ramos MV, Mejias MP, Sabbione F, Fernandez-Brando RJ, Santiago AP, Amaral MM, Exeni R, Trevani AS, Palermo MS (2016) Induction of neutrophil extracellular traps in Shiga toxin-associated hemolytic uremic syndrome. J Innate Immun 8:400–411
Brigotti M, Carnicelli D, Arfilli V, Tamassia N, Borsetti F, Fabbri E, Tazzari PL, Ricci F, Pagliaro P, Spisni E, Cassatella MA (2013) Identification of TLR4 as the receptor that recognizes Shiga toxins in human neutrophils. J Immunol 191:4748–4758
Kniep B, Monner DA, Schwulera U, Muhlradt PF (1985) Glycosphingolipids of the globo-series are associated with the monocytic lineage of human myeloid cells. Eur J Biochem 149:187–191
van Setten PA, Monnens LA, Verstraten RG, van den Heuvel LP, van Hinsbergh VW (1996) Effects of verocytotoxin-1 on nonadherent human monocytes: binding characteristics, protein synthesis, and induction of cytokine release. Blood 88:174–183
Tesh VL, Ramegowda B, Samuel JE (1994) Purified Shiga-like toxins induce expression of proinflammatory cytokines from murine peritoneal macrophages. Infect Immun 62:5085–5094
Lee SY, Cherla RP, Tesh VL (2007) Simultaneous induction of apoptotic and survival signaling pathways in macrophage-like THP-1 cells by Shiga toxin 1. Infect Immun 75:1291–1302
Brando RJ, Miliwebsky E, Bentancor L, Deza N, Baschkier A, Ramos MV, Fernandez GC, Meiss R, Rivas M, Palermo MS (2008) Renal damage and death in weaned mice after oral infection with Shiga toxin 2-producing Escherichia coli strains. Clin Exp Immunol 153:297–306
Cabrera G, Fernandez-Brando RJ, Abrey-Recalde MJ, Baschkier A, Pinto A, Goldstein J, Zotta E, Meiss R, Rivas M, Palermo MS (2014) Retinoid levels influence enterohemorrhagic Escherichia coli infection and Shiga toxin 2 susceptibility in mice. Infect Immun 82:3948–3957
Palermo M, Alves-Rosa F, Rubel C, Fernandez GC, Fernandez-Alonso G, Alberto F, Rivas M, Isturiz M (2000) Pretreatment of mice with lipopolysaccharide (LPS) or IL-1beta exerts dose-dependent opposite effects on Shiga toxin-2 lethality. Clin Exp Immunol 119:77–83
Stearns-Kurosawa DJ, SY O, Cherla RP, Lee MS, Tesh VL, Papin J, Henderson J, Kurosawa S (2013) Distinct renal pathology and a chemotactic phenotype after enterohemorrhagic Escherichia coli Shiga toxins in non-human primate models of hemolytic uremic syndrome. Am J Pathol 182:1227–1238
Mohawk KL, O’Brien AD (2011) Mouse models of Escherichia coli O157:H7 infection and Shiga toxin injection. J Biomed Biotechnol 2011:258185
Obata F, Tohyama K, Bonev AD, Kolling GL, Keepers TR, Gross LK, Nelson MT, Sato S, Obrig TG (2008) Shiga toxin 2 affects the central nervous system through receptor globotriaosylceramide localized to neurons. J Infect Dis 198:1398–1406
Takahashi K, Funata N, Ikuta F, Sato S (2008) Neuronal apoptosis and inflammatory responses in the central nervous system of a rabbit treated with Shiga toxin-2. J Neuroinflammation 5:11
Tironi-Farinati C, Geoghegan PA, Cangelosi A, Pinto A, Loidl CF, Goldstein J (2013) A translational murine model of sub-lethal intoxication with Shiga toxin 2 reveals novel ultrastructural findings in the brain striatum. PLoS One 8:e55812
Fernandez GC, Lopez MF, Gomez SA, Ramos MV, Bentancor LV, Fernandez-Brando RJ, Landoni VI, Dran GI, Meiss R, Isturiz MA, Palermo MS (2006) Relevance of neutrophils in the murine model of haemolytic uraemic syndrome: mechanisms involved in Shiga toxin type 2-induced neutrophilia. Clin Exp Immunol 146:76–84
Ibarra C, Amaral MM, Palermo MS (2013) Advances in pathogenesis and therapy of hemolytic uremic syndrome caused by Shiga toxin-2. IUBMB Life 65:827–835
Miyamoto Y, Iimura M, Kaper JB, Torres AG, Kagnoff MF (2006) Role of Shiga toxin versus H7 flagellin in enterohaemorrhagic Escherichia coli signalling of human colon epithelium in vivo. Cell Microbiol 8:869–879
Calderon Toledo C, Rogers TJ, Svensson M, Tati R, Fischer H, Svanborg C, Karpman D (2008) Shiga toxin-mediated disease in MyD88-deficient mice infected with Escherichia coli O157:H7. Am J Pathol 173:1428–1439
Gibson DL, Ma C, Bergstrom KS, Huang JT, Man C, Vallance BA (2008) MyD88 signalling plays a critical role in host defence by controlling pathogen burden and promoting epithelial cell homeostasis during Citrobacter rodentium-induced colitis. Cell Microbiol 10:618–631
Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R (2004) Recognition of commensal microflora by Toll-like receptors is required for intestinal homeostasis. Cell 118:229–241
Kramer S, Sellge G, Lorentz A, Krueger D, Schemann M, Feilhauer K, Gunzer F, Bischoff SC (2008) Selective activation of human intestinal mast cells by Escherichia coli hemolysin. J Immunol 181:1438–1445
Bell CJ, Elliott EJ, Wallace JL, Redmond DM, Payne J, Li Z, O’Loughlin EV (2000) Do eicosanoids cause colonic dysfunction in experimental E. coli O157:H7 (EHEC) infection? Gut 46:806–812
Cabrera G, Fernandez-Brando RJ, Mejias MP, Ramos MV, Abrey-Recalde MJ, Vanzulli S, Vermeulen M, Palermo MS (2015) Leukotriene C4 increases the susceptibility of adult mice to Shiga toxin-producing Escherichia coli infection. Int J Med Microbiol 305:910–917
Fernandez GC, Rubel C, Dran G, Gomez S, Isturiz MA, Palermo MS (2000) Shiga toxin-2 induces neutrophilia and neutrophil activation in a murine model of hemolytic uremic syndrome. Clin Immunol 95:227–234
Gomez SA, Fernandez GC, Camerano G, Dran G, Rosa FA, Barrionuevo P, Isturiz MA, Palermo MS (2005) Endogenous glucocorticoids modulate neutrophil function in a murine model of haemolytic uraemic syndrome. Clin Exp Immunol 139:65–73
Gomez SA, Fernandez GC, Vanzulli S, Dran G, Rubel C, Berki T, Isturiz MA, Palermo MS (2003) Endogenous glucocorticoids attenuate Shiga toxin-2-induced toxicity in a mouse model of haemolytic uraemic syndrome. Clin Exp Immunol 131:217–224
Gomez SA, Abrey-Recalde MJ, Panek CA, Ferrarotti NF, Repetto MG, Mejias MP, Fernandez GC, Vanzulli S, Isturiz MA, Palermo MS (2013) The oxidative stress induced in vivo by Shiga toxin-2 contributes to the pathogenicity of haemolytic uraemic syndrome. Clin Exp Immunol 173:463–472
Salvemini D, de Nucci G, Sneddon JM, Vane JR (1989) Superoxide anions enhance platelet adhesion and aggregation. Br J Pharmacol 97:1145–1150
Nath KA, Norby SM (2000) Reactive oxygen species and acute renal failure. Am J Med 109:665–678
Palermo MS, Alves Rosa MF, Van Rooijen N, Isturiz MA (1999) Depletion of liver and splenic macrophages reduces the lethality of Shiga toxin-2 in a mouse model. Clin Exp Immunol 116:462–467
Ramos MV, Auvynet C, Poupel L, Rodero M, Mejias MP, Panek CA, Vanzulli S, Combadiere C, Palermo M (2012) Chemokine receptor CCR1 disruption limits renal damage in a murine model of hemolytic uremic syndrome. Am J Pathol 180:1040–1048
Keepers TR, Gross LK, Obrig TG (2007) Monocyte chemoattractant protein 1, macrophage inflammatory protein 1 alpha, and RANTES recruit macrophages to the kidney in a mouse model of hemolytic-uremic syndrome. Infect Immun 75:1229–1236
Roche JK, Keepers TR, Gross LK, Seaner RM, Obrig TG (2007) CXCL1/KC and CXCL2/MIP-2 are critical effectors and potential targets for therapy of Escherichia coli O157:H7-associated renal inflammation. Am J Pathol 170:526–537
Obata F, Obrig T (2014) Role of Shiga/Vero toxins in pathogenesis. Microbiol Spectr. https://doi.org/10.1128/microbiolspec.EHEC-0005-2013
Lucero MS, Mirarchi F, Goldstein J, Silberstein C (2012) Intraperitoneal administration of Shiga toxin 2 induced neuronal alterations and reduced the expression levels of aquaporin 1 and aquaporin 4 in rat brain. Microb Pathog 53:87–94
Amran MY, Fujii J, Suzuki SO, Kolling GL, Villanueva SY, Kainuma M, Kobayashi H, Kameyama H, Yoshida S (2013) Investigation of encephalopathy caused by Shiga toxin 2c-producing Escherichia coli infection in mice. PLoS One 8:e58959
Isogai E, Isogai H, Kimura K, Hayashi S, Kubota T, Fujii N, Takeshi K (1998) Role of tumor necrosis factor alpha in gnotobiotic mice infected with an Escherichia coli O157:H7 strain. Infect Immun 66:197–202
Pradhan S, Pellino C, MacMaster K, Coyle D, Weiss AA (2016) Shiga toxin mediated neurologic changes in Murine model of disease. Front Cell Infect Microbiol 6:114
Porubsky S, Federico G, Muthing J, Jennemann R, Gretz N, Buttner S, Obermuller N, Jung O, Hauser IA, Grone E, Geiger H, Grone HJ, Betz C (2014) Direct acute tubular damage contributes to Shigatoxin-mediated kidney failure. J Pathol 234:120–133
Buteau C, Proulx F, Chaibou M, Raymond D, Clermont MJ, Mariscalco MM, Lebel MH, Seidman E (2000) Leukocytosis in children with Escherichia coli O157:H7 enteritis developing the hemolytic-uremic syndrome. Pediatr Infect Dis J 19:642–647
Fernandez GC, Gomez SA, Ramos MV, Bentancor LV, Fernandez-Brando RJ, Landoni VI, Lopez L, Ramirez F, Diaz M, Alduncin M, Grimoldi I, Exeni R, Isturiz MA, Palermo MS (2007) The functional state of neutrophils correlates with the severity of renal dysfunction in children with hemolytic uremic syndrome. Pediatr Res 61:123–128
Inward CD, Howie AJ, Fitzpatrick MM, Rafaat F, Milford DV, Taylor CM (1997) Renal histopathology in fatal cases of diarrhoea-associated haemolytic uraemic syndrome. British Association for Paediatric Nephrology. Pediatr Nephrol 11:556–559
van Setten PA, van Hinsbergh VW, van den Heuvel LP, Preyers F, Dijkman HB, Assmann KJ, van der Velden TJ, Monnens LA (1998) Monocyte chemoattractant protein-1 and interleukin-8 levels in urine and serum of patents with hemolytic uremic syndrome. Pediatr Res 43:759–767
Brigotti M, Tazzari PL, Ravanelli E, Carnicelli D, Barbieri S, Rocchi L, Arfilli V, Scavia G, Ricci F, Bontadini A, Alfieri RR, Petronini PG, Pecoraro C, Tozzi AE, Caprioli A (2010) Endothelial damage induced by Shiga toxins delivered by neutrophils during transmigration. J Leukoc Biol 88:201–210
Valles PG, Melechuck S, Gonzalez A, Manucha W, Bocanegra V, Valles R (2012) Toll-like receptor 4 expression on circulating leucocytes in hemolytic uremic syndrome. Pediatr Nephrol 27:407–415
Fernandez GC, Gomez SA, Rubel CJ, Bentancor LV, Barrionuevo P, Alduncin M, Grimoldi I, Exeni R, Isturiz MA, Palermo MS (2005) Impaired neutrophils in children with the typical form of hemolytic uremic syndrome. Pediatr Nephrol 20:1306–1314
Fernandez GC, Rubel C, Barrionuevo P, Lopez L, Ramirez F, Diaz M, Isturiz MA, Palermo MS (2002) Phenotype markers and function of neutrophils in children with hemolytic uremic syndrome. Pediatr Nephrol 17:337–344
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303:1532–1535
Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, Jr., Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD (2010) Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 107:15880-15885
Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, Grone HJ, Brinkmann V, Jenne DE (2009) Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med 15:623–625
Fuchs TA, Kremer Hovinga JA, Schatzberg D, Wagner DD, Lammle B (2012) Circulating DNA and myeloperoxidase indicate disease activity in patients with thrombotic microangiopathies. Blood 120:1157–1164
Leffler J, Prohaszka Z, Mikes B, Sinkovits G, Ciacma K, Farkas P, Reti M, Kelen K, Reusz GS, Szabo AJ, Martin M, Blom AM (2017) Decreased neutrophil extracellular trap degradation in Shiga toxin-associated haemolytic uraemic syndrome. J Innate Immun 9:12–21
von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Kollnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B, Massberg S (2012) Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med 209:819–835
Wang H, Wang C, Zhao MH, Chen M (2015) Neutrophil extracellular traps can activate alternative complement pathways. Clin Exp Immunol 181:518–527
Leffler J, Martin M, Gullstrand B, Tyden H, Lood C, Truedsson L, Bengtsson AA, Blom AM (2012) Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol 188:3522–3531
Morigi M, Galbusera M, Gastoldi S, Locatelli M, Buelli S, Pezzotta A, Pagani C, Noris M, Gobbi M, Stravalaci M, Rottoli D, Tedesco F, Remuzzi G, Zoja C (2011) Alternative pathway activation of complement by Shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J Immunol 187:172–180
Stahl AL, Sartz L, Karpman D (2011) Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood 117:5503–5513
Locatelli M, Buelli S, Pezzotta A, Corna D, Perico L, Tomasoni S, Rottoli D, Rizzo P, Conti D, Thurman JM, Remuzzi G, Zoja C, Morigi M (2014) Shiga toxin promotes podocyte injury in experimental hemolytic uremic syndrome via activation of the alternative pathway of complement. J Am Soc Nephrol 25:1786–1798
Freedman JE (2008) Oxidative stress and platelets. Arterioscler Thromb Vasc Biol 28:s11–s16
Perricone C, De Carolis C, Perricone R (2009) Glutathione: a key player in autoimmunity. Autoimmun Rev 8:697–701
Ferraris V, Acquier A, Ferraris JR, Vallejo G, Paz C, Mendez CF (2011) Oxidative stress status during the acute phase of haemolytic uraemic syndrome. Nephrol Dial Transplant 26:858–864
Aiassa V, Baronetti JL, Paez PL, Barnes AI, Albrecht C, Pellarin G, Eraso AJ, Albesa I (2011) Increased advanced oxidation of protein products and enhanced total antioxidant capacity in plasma by action of toxins of Escherichia coli STEC. Toxicol in Vitro 25:426–431
Li Volti S, Di Giacomo C, Garozzo R, Campisi A, Mollica F, Vanella A (1993) Impaired antioxidant defense mechanisms in two children with hemolytic-uremic syndrome. Ren Fail 15:523–528
Facorro G, Aguirre F, Florentin L, Diaz M, De Paoli T, Ihlo JE, Hager AA, Sanchez Avalos JC, Farach HA, Poole CP Jr (1997) Oxidative stress and membrane fluidity in erythrocytes from patients with hemolytic uremic syndrome. Acta Physiol Pharmacol Ther Latinoam 47:137–146
Dubey NK, Yadav P, Dutta AK, Kumar V, Ray GN, Batra S (2000) Free oxygen radicals in acute renal failure. Indian Pediatr 37:153–158
Powell HR, McCredie DA, Taylor CM, Burke JR, Walker RG (1984) Vitamin E treatment of haemolytic uraemic syndrome. Arch Dis Child 59:401–404
Fernandez GC, Ramos MV, Gomez SA, Dran GI, Exeni R, Alduncin M, Grimoldi I, Vallejo G, Elias-Costa C, Isturiz MA, Palermo MS (2005) Differential expression of function-related antigens on blood monocytes in children with hemolytic uremic syndrome. J Leukoc Biol 78:853–861
Fernandez GC, Ramos MV, Landoni VI, Bentancor LV, Fernandez-Brando RJ, Exeni R, Laso Mdel C, Exeni A, Grimoldi I, Isturiz MA, Palermo MS (2012) Cytokine production is altered in monocytes from children with hemolytic uremic syndrome. J Clin Immunol 32:622–631
Ramos MV, Ruggieri M, Panek AC, Mejias MP, Fernandez-Brando RJ, Abrey-Recalde MJ, Exeni A, Barilari C, Exeni R, Palermo MS (2015) Association of haemolytic uraemic syndrome with dysregulation of chemokine receptor expression in circulating monocytes. Clin Sci (Lond) 129:235–244
Ramos MV, Fernandez GC, Patey N, Schierloh P, Exeni R, Grimoldi I, Vallejo G, Elias-Costa C, Del Carmen Sasiain M, Trachtman H, Combadiere C, Proulx F, Palermo MS (2007) Involvement of the fractalkine pathway in the pathogenesis of childhood hemolytic uremic syndrome. Blood 109:2438–2445
Proulx F, Toledano B, Phan V, Clermont MJ, Mariscalco MM, Seidman EG (2002) Circulating granulocyte colony-stimulating factor, C-X-C, and C-C chemokines in children with Escherichia coli O157:H7 associated hemolytic uremic syndrome. Pediatr Res 52:928–934
Proulx F, Litalien C, Turgeon JP, Mariscalco MM, Seidman E (2000) Circulating levels of transforming growth factor-beta1 and lymphokines among children with hemolytic uremic syndrome. Am J Kidney Dis 35:29–34
Shimizu M, Kuroda M, Inoue N, Konishi M, Igarashi N, Taneichi H, Kanegane H, Ito M, Saito S, Yachie A (2014) Extensive serum biomarker analysis in patients with enterohemorrhagic Escherichia coli O111-induced hemolytic-uremic syndrome. Cytokine 66:1–6
Shiraishi M, Ichiyama T, Matsushige T, Iwaki T, Iyoda K, Fukuda K, Makata H, Matsubara T, Furukawa S (2008) Soluble tumor necrosis factor. Receptor 1 and tissue inhibitor of metalloproteinase-1 in hemolytic uremic syndrome with encephalopathy. J Neuroimmunol 196:147–152
Murata A, Shimazu T, Yamamoto T, Taenaka N, Nagayama K, Honda T, Sugimoto H, Monden M, Matsuura N, Okada S (1998) Profiles of circulating inflammatory- and anti-inflammatory cytokines in patients with hemolytic uremic syndrome due to E. coli O157 infection. Cytokine 10:544–548
Page AV, Tarr PI, Watkins SL, Rajwans N, Petruzziello-Pellegrini TN, Marsden PA, Kain KC, Liles WC (2013) Dysregulation of angiopoietin 1 and 2 in Escherichia coli O157:H7 infection and the hemolytic-uremic syndrome. J Infect Dis 208:929–933
Siegler RL, Edwin SS, Christofferson RD, Mitchell MD (1991) Endothelin in the urine of children with the hemolytic uremic syndrome. Pediatrics 88:1063–1066
Yamamoto T, Nagayama K, Satomura K, Honda T, Okada S (2000) Increased serum IL-10 and endothelin levels in hemolytic uremic syndrome caused by Escherichia coli O157. Nephron 84:326–332
Decaluwe H, Harrison LM, Mariscalco MM, Gendrel D, Bohuon C, Tesh VL, Proulx F (2006) Procalcitonin in children with Escherichia coli O157:H7 associated hemolytic uremic syndrome. Pediatr Res 59:579–583
Robson WL, Leung AK, Fick GH, McKenna AI (1992) Hypocomplementemia and leukocytosis in diarrhea-associated hemolytic uremic syndrome. Nephron 62:296–299
Thurman JM, Marians R, Emlen W, Wood S, Smith C, Akana H, Holers VM, Lesser M, Kline M, Hoffman C, Christen E, Trachtman H (2009) Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol 4:1920–1924
Ferraris JR, Ferraris V, Acquier AB, Sorroche PB, Saez MS, Ginaca A, Mendez CF (2015) Activation of the alternative pathway of complement during the acute phase of typical haemolytic uraemic syndrome. Clin Exp Immunol 181:118–125
Arvidsson I, Stahl AL, Hedstrom MM, Kristoffersson AC, Rylander C, Westman JS, Storry JR, Olsson ML, Karpman D (2015) Shiga toxin-induced complement-mediated hemolysis and release of complement-coated red blood cell-derived microvesicles in hemolytic uremic syndrome. J Immunol 194:2309–2318
Ahlenstiel-Grunow T, Hachmeister S, Bange FC, Wehling C, Kirschfink M, Bergmann C, Pape L (2016) Systemic complement activation and complement gene analysis in enterohaemorrhagic Escherichia coli-associated paediatric haemolytic uraemic syndrome. Nephrol Dial Transplant 31:1114–1121
Orth-Holler D, Wurzner R (2014) Role of complement in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Semin Thromb Hemost 40:503–507
Rosales A, Hofer J, Zimmerhackl LB, Jungraithmayr TC, Riedl M, Giner T, Strasak A, Orth-Holler D, Wurzner R, Karch H (2012) Need for long-term follow-up in enterohemorrhagic Escherichia coli-associated hemolytic uremic syndrome due to late-emerging sequelae. Clin Infect Dis 54:1413–1421
Exeni RA (2006) Hemolytic uremic syndrome. Clinical manifestations. Treatment. Medicina (B Aires) 66(Suppl 3):6–10
Magnus T, Rother J, Simova O, Meier-Cillien M, Repenthin J, Moller F, Gbadamosi J, Panzer U, Wengenroth M, Hagel C, Kluge S, Stahl RK, Wegscheider K, Urban P, Eckert B, Glatzel M, Fiehler J, Gerloff C (2012) The neurological syndrome in adults during the 2011 northern German E. coli serotype O104:H4 outbreak. Brain 135:1850–1859
Menne J, Nitschke M, Stingele R, Abu-Tair M, Beneke J, Bramstedt J, Bremer JP, Brunkhorst R, Busch V, Dengler R, Deuschl G, Fellermann K, Fickenscher H, Gerigk C, Goettsche A, Greeve J, Hafer C, Hagenmuller F, Haller H, Herget-Rosenthal S, Hertenstein B, Hofmann C, Lang M, Kielstein JT, Klostermeier UC, Knobloch J, Kuehbacher M, Kunzendorf U, Lehnert H, Manns MP, Menne TF, Meyer TN, Michael C, Munte T, Neumann-Grutzeck C, Nuernberger J, Pavenstaedt H, Ramazan L, Renders L, Repenthin J, Ries W, Rohr A, Rump LC, Samuelsson O, Sayk F, Schmidt BM, Schnatter S, Schocklmann H, Schreiber S, von Seydewitz CU, Steinhoff J, Stracke S, Suerbaum S, van de Loo A, Vischedyk M, Weissenborn K, Wellhoner P, Wiesner M, Zeissig S, Buning J, Schiffer M, Kuehbacher T (2012) Validation of treatment strategies for enterohaemorrhagic Escherichia coli O104:H4 induced haemolytic uraemic syndrome: case-control study. BMJ 345:e4565
Delmas Y, Vendrely B, Clouzeau B, Bachir H, Bui HN, Lacraz A, Helou S, Bordes C, Reffet A, Llanas B, Skopinski S, Rolland P, Gruson D, Combe C (2014) Outbreak of Escherichia coli O104:H4 haemolytic uraemic syndrome in France: outcome with eculizumab. Nephrol Dial Transplant 29:565–572
Saini A, Emke AR, Silva MC, Perlman SJ (2015) Response to Eculizumab in Escherichia coli O157:H7-induced hemolytic uremic syndrome with severe neurological manifestations. Clin Pediatr (Phila) 54:387–389
Pape L, Hartmann H, Bange FC, Suerbaum S, Bueltmann E, Ahlenstiel-Grunow T (2015) Eculizumab in typical hemolytic uremic syndrome (HUS) with neurological involvement. Medicine (Baltimore) 94:e1000
Lapeyraque AL, Malina M, Fremeaux-Bacchi V, Boppel T, Kirschfink M, Oualha M, Proulx F, Clermont MJ, Le Deist F, Niaudet P, Schaefer F (2011) Eculizumab in severe Shiga-Toxin-Associated HUS. N Engl J Med 364:2561–2563
Takanashi J, Taneichi H, Misaki T, Yahata Y, Okumura A, Ishida Y, Miyawaki T, Okabe N, Sata T, Mizuguchi M (2014) Clinical and radiologic features of encephalopathy during 2011 E. coli O111 outbreak in Japan. Neurology 82:564–572
Valles PG, Pesle S, Piovano L, Davila E, Peralta M, Principi I, Lo Giudice P (2005) Postdiarrheal Shiga toxin-mediated hemolytic uremic syndrome similar to septic shock. Medicina (B Aires) 65:395–401
Igarashi T, Ito S, Sako M, Saitoh A, Hataya H, Mizuguchi M, Morishima T, Ohnishi K, Kawamura N, Kitayama H, Ashida A, Kaname S, Taneichi H, Tang J, Ohnishi M (2014) Guidelines for the management and investigation of hemolytic uremic syndrome. Clin Exp Nephrol 18:525–557
Hagel C, Krasemann S, Loffler J, Puschel K, Magnus T, Glatzel M (2015) Upregulation of Shiga toxin receptor CD77/Gb3 and interleukin-1beta expression in the brain of EHEC patients with hemolytic uremic syndrome and neurologic symptoms. Brain Pathol 25:146–156
Kuroda M, Shimizu M, Inoue N, Ikeno I, Nakagawa H, Yokoi A, Niida Y, Konishi M, Kaneda H, Igarashi N, Yamahana J, Taneichi H, Kanegane H, Ito M, Saito S, Furuichi K, Wada T, Nakagawa M, Yokoyama H, Yachie A (2015) Serum tau protein as a marker of disease activity in enterohemorrhagic Escherichia coli O111-induced hemolytic uremic syndrome. Neurochem Int 85–86:24–30
Exeni RA, Fernandez GC, Palermo MS (2007) Role of polymorphonuclear leukocytes in the pathophysiology of typical hemolytic uremic syndrome. Sci World J 7:1155–1164
Acknowledgements
This work was supported by a grant from Agencia Nacional de Promoción Científica y Tecnológica, Argentina (PIDC 2014/0020).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Answers to questions:
1. a; 2. c; 3. d; 4. a; 5. b
Rights and permissions
About this article

Cite this article
Exeni, R.A., Fernandez-Brando, R.J., Santiago, A.P. et al. Pathogenic role of inflammatory response during Shiga toxin-associated hemolytic uremic syndrome (HUS). Pediatr Nephrol 33, 2057–2071 (2018). https://doi.org/10.1007/s00467-017-3876-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-017-3876-0