
Abstract
Acute kidney injury is a common and serious complication after congenital heart surgery, particularly among infants. This comorbidity has been independently associated with adverse outcomes including an increase in mortality. Postoperative acute kidney injury has a complex pathophysiology with many risk factors, and therefore no single medication or therapy has been demonstrated to be effective for treatment or prevention. However, it has been established that the associated fluid overload is one of the major determinants of morbidity, particularly in infants after cardiac surgery. Therefore, in the absence of an intervention to prevent acute kidney injury, much of the effort to improve outcomes has focused on treating and preventing fluid overload. Early renal replacement therapy, often in the form of peritoneal dialysis, has been shown to be safe and beneficial in infants with oliguria after heart surgery. As understanding of the pathophysiology of acute kidney injury and the ability to confidently diagnose it earlier continues to evolve, it is likely that novel preventative and therapeutic interventions will be available in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute kidney injury (AKI) is a frequent and serious complication after pediatric cardiovascular surgery affecting up to 30–60% of patients [1–3]. Surgical palliation of congenital heart disease is the most common cause of AKI among children [4]. It is well established that the development of postoperative AKI is an independent risk factor for worse outcomes, including mortality [1–3, 5, 6]. Recently, there has been a great deal of research focused on the earlier diagnosis of AKI with many promising novel biomarkers; however, successful therapies lag behind. This review focuses on the diagnosis and management of AKI and its sequelae, fluid overload, within patients with congenital heart disease. As AKI occurs mainly in infants after cardiac surgery with a cardiopulmonary bypass (CPB), this cohort is the focus of most published literature and likewise this review highlights this population.
Definitions of acute kidney injury
Historically, the reported incidence of AKI after congenital heart surgery has varied, in large part because of the lack of a consistent definition. For example, a systematic review of 28 studies between 1965 and 1989 of postoperative AKI found that no two studies used the same diagnostic criteria for AKI [7]. In an effort at standardization, definitions of kidney injury have been created for research and clinical utility and have been modified for pediatric cohorts, most notably the Acute Kidney Injury Network (AKIN), Risk Injury Failure Loss End-stage renal disease (RIFLE), and pediatric modified Risk Injury Failure Loss End-stage renal disease (pRIFLE) classifications (Table 1) [8–10]. However, the most recent and agreed upon consensus definition is that described by the Kidney Disease: Improving Global Outcomes (KDIGO) group. These criteria have three stages and are defined by urine output and creatinine criteria (Table 1). A comparison of AKIN, pRIFLE, and KDIGO classifications in pediatric patients after heart surgery found that all systems were sensitive for AKI and found associations with mortality; however, pRIFLE classification was most sensitive at detecting AKI in the infant group [11]. Because of logistical challenges with the collection and quantification of urine in the pediatric and particularly infant populations, urine output criteria have been infrequently used within research studies and therefore are less well validated than the serum creatinine criteria. However, it has been suggested that urine output criteria are more sensitive for the diagnosis of AKI in pediatric critically ill patients [12].
Diagnosis of AKI
One of the main challenges to the management of AKI after CPB is the delay in accurate diagnosis. In current clinical practice, laboratory evidence of AKI continues to rely on serum creatinine elevation, a functional biomarker that is insensitive to renal tubule injury. Creatinine elevation is not seen until 50% of kidney function is lost and often takes 24–48 h after the initial insult to elevate [1, 2, 13]. Waiting for creatinine elevation to modify treatment is analogous to waiting for cardiac output to decrease to begin the management of a myocardial infarction. The use of biomarkers of structural injury allows the diagnosis to occur before decreased glomerular filtration causes impairment in urine output, and before the late finding of creatinine elevation (Fig. 1).

In the progression from initial renal insult to kidney failure, biomarkers of renal function only show elevation late after the development of kidney injury, and the clinical manifestation of oliguria occurs after glomerular filtration is impaired. Biomarkers of structural kidney injury diagnose kidney injury early, before glomerular filtration is affected
Like creatinine, serum cystatin-C is a functional biomarker that demonstrates elevation after AKI. It has been demonstrated that cystatin-C can be used as a functional biomarker of injury and diagnose AKI sooner than creatinine with improved specificity and sensitivity [14, 15]. Serum cystatin-C is also used by some centers for the estimation of glomerular filtration rate, as it has been demonstrated to be a more precise measure of GFR and unlike creatinine is not affected by muscle mass [16].
Unlike myocardial infarction, in pediatrics there are currently no FDA-approved troponin-like biomarkers for diagnosing renal structural injury early enough to direct care. However, the FDA has recently approved the NephroCheck test for the prediction of AKI in adults. The NephroCheck test is a urine-based test measuring insulin-like growth factor-binding protein 7 (IFGBP-7) and tissue inhibitor of metalloproteinases-2 (TIMP-2), which are inducers of cell cycle arrest, an important mechanism in the progression of AKI. The test quantifies the product of these biomarkers to predict the likelihood of developing AKI. In adults, the NephroCheck test has been demonstrated to predict AKI as early as 4 h post bypass and to accurately predict renal recovery [17]. Although not yet validated in pediatric populations, this biomarker shows promise of early diagnosis [18].
This review does not address other novel biomarkers that are being used exclusively in research settings [19–21]. It is likely that the future of AKI diagnosis will be aided by these novel biomarkers. Given the complexity of the pathophysiology of AKI, diagnosis may be optimized by using panels of biomarkers that each detects different pathways of injury, often with different temporality. In addition, the use of biomarker panels may help to illustrate the specific pathophysiology of injury, allowing the modification of management [5]. As a precursor to this concept, Basu and colleagues demonstrated that combining the functional biomarker cystatin-C and the tubular injury biomarker NGAL could increase the sensitivity and specificity of the diagnosis of AKI in children after CPB compared with creatinine alone [22].
Pathophysiology of AKI
The mechanism of AKI after CPB is multifactorial, including renal ischemia and reperfusion injury, a maladaptive inflammatory response, oxidative stress, microemboli, and alterations in tubule cell metabolism. Given the complexity of renal injury pathophysiology and the multi-modal mechanism, the difficulty in developing a single therapeutic modality becomes more apparent.
All organs, including the kidney, have the ability to maintain consistent perfusion during a period of diminished blood flow through a process called auto-regulation. Neurohormonal pathways modulate the constriction and vasodilation of afferent and efferent arterioles to titrate vascular resistance to maintain a relatively consistent perfusion pressure. The ability of the kidney to adapt to major hemodynamic changes during the intraoperative and postoperative periods, such as episodes of hypotension and/or elevation in central venous pressure, is unknown, especially in pediatric and neonatal patients. Furthermore, auto-regulation may be inhibited by the administration of vasoactive and steroid medications and a proinflammatory state [23].
Inflammation plays a role in most types of ischemic and reperfusion renal injury [24]. However, the role is even greater among cardiac surgical patients owing to direct tissue and endothelial injury and cellular contact with the artificial surface of the CPB circuit [25]. Inflammatory cascades integral in the development of injury include the proinflammatory cytokine tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), interleukin-1β (IL-1β), and transforming growth factor-β (TGF-β). The consistent up-regulation of these cytokines makes them obvious targets for diagnosis and therapies.
Furthermore, CPB exposes blood cells to non-physiological surfaces and shear forces, leading to cell lysis and release of plasma-free hemoglobin into the circulation. Microemboli composed of fibrin, platelets, cellular debris, fat, and air are formed during cardiac surgery. Although larger emboli are filtered by the CPB system, smaller emboli may be circulated to the renal capillary bed and directly cause ischemia and injury [26]. Activation of the systemic inflammatory response further potentiates cell damage via oxidative stress injury and coagulopathy.
On a cellular level, ischemic injury leads to profound ATP depletion and induction of nitric oxide synthetase [24]. A number of oxidative and cell death mechanisms are then induced, including activation of caspase, alteration in intracellular calcium, and generation of reactive oxygen molecules. These pathways progress, resulting primarily in apoptosis, alterations in tubule structure, and oxidant injury.
Risk factors for AKI
Although risk factors for the development of AKI have been identified, few of them are modifiable. Younger age at repair, higher surgical complexity, longer bypass, preoperative ventilation, and the use of deep hypothermic circulatory arrest have been shown to be risk factors, but also correlate with a more critical patient substrate in whom surgery often cannot be delayed or modified [1, 3, 13]. In the TRIBE-AKI consortium, bypass time was found to be a strong risk factor for the development of AKI. After controlling for age and surgical severity, bypass times of 120–180 min had an odds ratio of 3.2 and bypass times of >180 min had an odds ratio of 7.6 for the development of AKI. When looking specifically at modifiable risk factors, the TRIBE-AKI consortium noted that 87% of patients with AKI had intraoperative hypotension, 15% were exposed to gentamicin, 56% were exposed to non-steroidal anti-inflammatory drugs (NSAIDs), and 6% experienced low cardiac output syndrome [1]. Lesions with a high postoperative central venous pressure have also been demonstrated to have a high incidence of AKI. These include lesions with residual right ventricular hypertrophy and diastolic dysfunction, such as repair of tetralogy of Fallot, and lesions with obligate central venous pressure elevation due to surgical palliation and postoperative physiology, such as Fontan surgery [27].
Given the same set of risk factors and a similar renal insult, some patients develop kidney injury and others do not. Although this may be influenced by chance or differences in surgical or bypass techniques, there is growing evidence that there may be selected gene variations that either predispose patients to or protect them from the development of AKI. Given the common mechanisms of postoperative AKI, genetic polymorphisms that are associated with renal inflammation, oxidative stress or vasoconstrictor response have been of greatest interest [28, 29]. A study among adult patients undergoing aortic-coronary surgery found that there were two alleles (interleukin 6–572C and angiotensinogen 842C) that showed a strong association with AKI in Caucasians [28]. The ability to predict AKI increased 4-fold when the use of genetic polymorphisms was added to clinical risk factors alone. It is likely that further polymorphisms will be detected in the future, the discovery of which may aid clinicians in defining higher risk populations and in developing targeted therapies.
Outcomes after AKI
Acute kidney injury after CPB is frequently a self-limited complication, often occurring in the first 24–48 h after surgery. In the TRIBE-AKI consortium almost half of patients met AKI diagnostic criteria for just 1 day and only 1 out of 9 patients continued to meet the definition by the fourth postoperative day [1]. For this reason, the importance of postoperative AKI has historically been minimized. However, recent research has emphasized the strong association of even minor degrees of AKI with worse outcomes.
Acute kidney injury is an independent risk factor for prolonged duration of mechanical ventilation, longer ICU and hospital stays, and mortality [1–3]. The TRIBE-AKI consortium found that 30% of patients with AKI were mechanically ventilated at 48 h postoperatively, as opposed to 8% of those without AKI [1]. Blinder and colleagues studied 430 infants after bypass and found that 52% developed AKI. A doubling of creatinine was associated with an odds ratio of death of 5.1, and tripling was associated with an odds ratio of 9.5. In this study, AKI was a stronger predictor of death than having single-ventricle physiology, or needing mechanical circulatory support [2].
Even small increases in creatinine are important. Compared with those with no change in creatinine, adults after cardiac surgery with a creatinine increase of just 0.1 to 0.5 mg/dl had a three-fold increase in the rate of mortality, and this association worsened with larger creatinine changes [30]. There are data among pediatric patients after cardiac surgery that even small changes in creatinine predict later development of more severe AKI [13].
The long-term impact of an isolated episode of AKI continues to be evaluated; however, it is likely not as benign as previously thought. Morgan et al. followed a cohort of neonates after cardiac surgery and found that 2–4 years postoperatively, children who developed AKI were at a higher risk of growth impairment, cardiac-related hospitalization, and increased health care utilization, even when controlling for gestational age, surgical type, preoperative ventilation, lactate elevation, and use of mechanical circulatory support [3]. Furthermore, although creatinine levels typically normalize in patients before discharge, there is evidence that those affected by AKI have persistent elevation of kidney injury biomarkers up to 7 years postoperatively [31]. It is unknown if these children will later develop clinical evidence for chronic kidney injury as they get older, but this argues against complete recovery.
Fluid overload
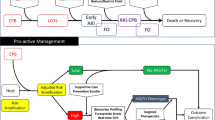
It had previously been hypothesized that AKI is a marker of poor perioperative condition and hemodynamic instability and that AKI itself does not lead to morbidity and mortality, but is just a bystander while poor cardiac output affects outcomes [32]. This notion has lost favorability as research has stressed the independent association of AKI with morbidity. The primary mechanism by which AKI causes worse outcomes is believed to be the development of fluid overload. Renal tubule injury leads to a decrease in glomerular filtration, which causes retention of free water among the other retained filtrate. CPB induces ischemia, inflammation, and capillary leak, and can lead to impaired cardiac output. Clinically, this appears as hypotension and often necessitates fluid resuscitation or administration of blood products. Low cardiac output activates a number of neurohormonal pathways, including the renin–angiotensin system, which collectively leads to fluid and salt retention. Fluid overload in the face of impaired cardiac output leads to elevated central venous pressure. At the same time, renal vascular resistance increases. The combination of all these factors leads to a further decline in the renal perfusion pressure and progressive kidney injury, which then leads to more fluid overload and the cycle continues (Fig. 2).

The relationship between acute kidney injury after cardiac surgery and fluid overload is complex. Common pathways of ischemia, inflammation, and low cardiac output cause both kidney injury and fluid overload. The resulting elevation of central venous pressure and renal vascular resistance worsens renal perfusion. Without intervention, this may precipitate further kidney injury, which then leads to more fluid overload, and the cycle continues
In addition to the swollen appearance of a patient with fluid overload, edema of various end organs occurs, with subsequent pulmonary edema, ascites, pleural effusions, myocardial edema, and gut and body wall edema. These edematous organs all individually become dysfunctional; pulmonary gas exchange is impaired and lung compliance decreases, swollen bowel walls absorb less nutrition and have impaired motility leading to feeding intolerance, skin breakdown leads to pressure ulcers and infection at catheter sites, ascites impairs cardiac preload and causes abdominal tamponade of peritoneal organs, and cardiac myocardium is less contractile with diastolic dysfunction, decreased preload, and impaired cardiac output. Among the other morbid effects, the decline in cardiac output and renal perfusion causes further renal dysfunction, leading to even more fluid overload. Without the ability to break this cycle, AKI may quickly become a major contributor of morbidity and mortality.
The association of fluid overload with mortality in critically ill children was first described by Sutherland, who compared the outcomes of patients with various levels of fluid overload at the time of initiation of dialysis [33]. He found that mortality increased directly with degree of fluid overload and that patients with <10% overload had a mortality of approximately 30%, while almost two-thirds of patients with >20% overload died during that admission. Specifically among neonates and children after cardiac surgery, those with fluid overload have longer ICU admissions, increased inotrope use, prolonged respiratory failure, increased rates of mechanical circulatory support, and cardiac arrest [34, 35].
Management of fluid overload
As there are no validated medications to treat AKI, the focus of management often shifts to treatment of fluid overload. Without a doubt, the quintessential management for fluid overload is restriction of fluid input, but this is often easier said than done. A common postoperative total fluid allowance for an infant after surgery is two-thirds of the maintenance dose. However, the challenge of management can be illustrated in the example of a critically ill 3-kg patient who is restricted to 8 ml per hour. This volume is often exceeded by vasoactive infusions, continuous sedation medications, diuretics, antibiotics, electrolyte replacements, and line flushes, even if adequate nutrition is withheld. Furthermore, if the infant is oliguric, with less than 1 ml of urine per kilogram per hour (a frequent occurrence), even the most modest fluid inputs will lead to fluid overload.
Pharmacological interventions intended to improve urine output include diuretics, renal vasodilators, and medications to improve cardiac output. Renal vasodilators and medications to improve cardiac output are theorized to improve glomerular perfusion, therefore improving urine output and potentially mitigating AKI. Retrospective studies of fenoldopam and aminophylline have suggested an improvement in urine output in neonates after cardiac surgery [36, 37]. However, none of the investigated renal vasodilators, including aminophylline, fenoldopam, and nesiritide, have been shown to decrease the incidence of AKI or augment urine output in randomized controlled trials in pediatric patients after cardiac surgery [38–40]. The use of diuretics in critically ill patients with AKI is ubiquitous; however, they are often ineffective and at worse, may even be detrimental, associated in some studies with a higher risk of mortality [41, 42]. Almost all diuretics must reach the tubular lumen by glomerular filtration or proximal tubular secretion to exert their action [43]. If AKI causes a decrease in glomerular filtration, diuretic delivery is impeded and these medications are thus less effective.
As medical interventions are often ineffective at preventing and treating fluid overload, early renal replacement therapy is becoming increasingly used in pediatric patients after cardiac surgery, in many cases to avoid the onset of fluid overload. The most common form of renal replacement therapy among patients with postoperative AKI is peritoneal dialysis (PD). Although some studies have reported an association with higher rates of mortality in patients who have received PD [44, 45], other studies note that historically, PD has often been used too rarely and too late [46, 47]. In addition to preventing the edema associated with multi-organ dysfunction, the use of PD may allow increased administration of fluids to provide adequate nutrition, essential blood products, and beneficial medications. It has also been suggested that PD might help to clear maladaptive cytokines that may propagate the inflammatory process [48]. In critically ill patients without cardiac disease, it is established that mortality is significantly lower if dialysis is commenced before significant fluid overload [33, 49]. However, it has only recently been demonstrated that children with fluid overload after cardiac surgery who go on to require renal replacement therapy have improved mortality if PD is performed earlier [46].
Reports of the systematic use of PD as an adjunct to postoperative fluid overload were published decades ago, and countless studies have since reported safety of the use [46–48, 50–53]. Among patients with fluid overload, the benefits are clear; however, the benefit of this invasive procedure to prevent fluid overload in infants with oliguria after cardiac surgery has only recently been established. A case-matched cohort study comparing patients with a peritoneal dialysis catheter placed at the time of surgery to others of a similar age and surgical procedure demonstrated that those with a peritoneal drainage catheter were less likely to develop fluid overload, had shorter durations of mechanical ventilation, shorter ICU stay and did not have an increase in expenditure [47]. A follow-up study of 73 infants with oliguria after cardiac surgery randomized patients to receive standardized doses of furosemide or PD [54]. Patients who received furosemide were three times more likely to develop fluid overload and were more likely to have prolonged mechanical ventilation and electrolyte abnormalities than those randomized to early PD. Importantly, as with most other studies, those with PD did not have any significant adverse outcomes, including peritonitis, bowel injury or significant hemodynamic instability.
To address concerns that the use of PD may adversely affect kidney function recovery, the Baylor group performed a randomized study among 20 patients receiving PD after pediatric cardiac surgery [55]. At the time of readiness to discontinue PD, patients were randomized to either stop dialysis or continue for another 24 h. There were no differences in the biomarkers of kidney injury among the groups, indicating that further tubular injury from the prolongation of dialysis was unlikely.
In patients who have been initiated on PD, several criteria for duration have been suggested. Most commonly, PD is discontinued as urine output increases. Because this is often difficult to discern, a furosemide challenge has been proposed, in which a 1-mg/kg dose of intravenous furosemide is given and urine output greater than 1 ml/kg/h for 4 h is considered an indication for the cessation of PD [55].
Intermittent hemodialysis (HD) and continuous venovenous hemofiltration (CVVH) are also options for fluid removal after surgery, and are commonly used when patients have other indications for dialysis, such as azotemia, hyperkalemia, or other electrolyte imbalances. These methods of dialysis deliver reliable and adjustable fluid removal and have several strengths and challenges compared with PD. Hemodialysis does not require abdominal installation of fluid and is therefore preferred in anyone who has had recent abdominal surgery, or cardiac surgery with peritoneal extension (vascular access device, pacemaker, etc.). Although the vascular access necessary to provide adequate hemofiltration has decreased in size, typically a 7-Fr catheter is the minimum-sized catheter for adequate blood flow. Larger catheters in larger patients are associated with improved dialysis performance and should be used as appropriate [8]. Dialysis catheter placement ideally utilizes the internal jugular vein, and may provide a challenge in infants and small children who may have a history of vascular access or vascular injury [8]. Intermittent HD allows patients to have therapy for several hours a day allowing for time disengaged from the circuit to provide comfort and mobility for rehabilitation.
Continuous renal replacement therapy, most typically via CVVH, offers several benefits over intermittent hemodialysis. The slow filtration afforded by this method avoids the rapid fluid shifts associated with intermittent HD, which may cause hemodynamic instability. This is especially important in smaller children and patients early after surgery. Similarly, a slower catheter flow rate allows the use of smaller catheters relative to HD. The CVVH circuit can easily be used in conjunction with extracorporeal membrane oxygenation [56]. Regardless of the modality of renal replacement therapy, early initiation of therapy is paramount.
Conclusion
Over the past 20 years, recognition of the importance of AKI after CPB in children has evolved thought paradigms, changing AKI from a typical perioperative stage in recovery to an epidemic that demands intervention. The ability and consistency by which AKI is diagnosed has greatly improved, but reliance on serum creatinine still hampers early diagnosis. Currently, there are no medications available to prevent or treat AKI and much of the management is focused on preventing and treating fluid overload. It is our hope that during the next 20 years we will see the diagnostic ability to identify the mechanism of injury earlier, allowing tailored interventions to be delivered in a timely manner. Only then will we see true improvement in one of the most significant comorbidities of patients with congenital heart disease.
References
Li S, Krawczeski CD, Zappitelli M, Devarajan P, Thiessen-Philbrook H, Coca SG, Kim RW, Parikh CR (2011) Incidence, risk factors, and outcomes of acute kidney injury after pediatric cardiac surgery—a prospective multicenter study. Crit Care Med 39:1493
Blinder JJ, Goldstein SL, Lee V-V, Baycroft A, Fraser CD, Nelson D, Jefferies JL (2012) Congenital heart surgery in infants: effects of acute kidney injury on outcomes. J Thorac Cardiovasc Surg 143:368–374
Morgan CJ, Zappitelli M, Robertson CM, Alton GY, Sauve RS, Joffe AR, Ross DB, Rebeyka IM, Western Canadian Complex Pediatric Therapies Follow-Up Group (2013) Risk factors for and outcomes of acute kidney injury in neonates undergoing complex cardiac surgery. J Pediatr 162:120–127.e1
Williams DM, Sreedhar SS, Mickell JJ, Chan JC (2002) Acute kidney failure: a pediatric experience over 20 years. Arch Pediatr Adolesc Med 156:893–900
Krawczeski CD, Goldstein SL, Woo JG, Wang Y, Piyaphanee N, Ma Q, Bennett M, Devarajan P (2011) Temporal relationship and predictive value of urinary acute kidney injury biomarkers after pediatric cardiopulmonary bypass. J Am Coll Cardiol 58:2301–2309
Machado MN, Nakazone MA, Maia LN (2014) Acute kidney injury based on KDIGO (Kidney Disease Improving Global Outcomes) criteria in patients with elevated baseline serum creatinine undergoing cardiac surgery. Rev Bras Cir Cardiovasc 29:299–307
Novis BK, Roizen MF, Aronson S, Thisted RA (1994) Association of preoperative risk factors with postoperative acute renal failure. Anesth Analg 78:143–149
Kellum JA, Lameire N (2013) Diagnosis, evaluation, and management of acute kidney injury: a KDIGO summary (Part 1). Crit Care 17:204
Akcan-Arikan A, Zappitelli M, Loftis L, Washburn K, Jefferson L, Goldstein S (2007) Modified RIFLE criteria in critically ill children with acute kidney injury. Kidney Int 71:1028–1035
Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, Levin A (2007) Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care 11:R31
Lex DJ, Tóth R, Cserép Z, Alexander SI, Breuer T, Sápi E, Szatmári A, Székely E, Gál J, Székely A (2014) A comparison of the systems for the identification of postoperative acute kidney injury in pediatric cardiac patients. Ann Thorac Surg 97:202–210
Kaddourah A, Basu RK, Bagshaw SM, Goldstein SL (2017) Epidemiology of acute kidney injury in critically ill children and young adults. N Engl J Med 376:11–20
Zappitelli M, Bernier P-L, Saczkowski RS, Tchervenkov CI, Gottesman R, Dancea A, Hyder A, Alkandari O (2009) A small post-operative rise in serum creatinine predicts acute kidney injury in children undergoing cardiac surgery. Kidney Int 76:885–892
Hassinger AB, Backer CL, Lane JC, Haymond S, Wang D, Wald EL (2012) Predictive power of serum cystatin C to detect acute kidney injury and pediatric-modified RIFLE class in children undergoing cardiac surgery. Pediatr Crit Care Med 13:435–440
Spahillari A, Parikh CR, Sint K, Koyner JL, Patel UD, Edelstein CL, Passik CS, Thiessen-Philbrook H, Swaminathan M, Shlipak MG (2012) Serum cystatin C- versus creatinine-based definitions of acute kidney injury following cardiac surgery: a prospective cohort study. Am J Kidney Dis 60:922–929
Dharnidharka VR, Kwon C, Stevens G (2002) Serum cystatin C is superior to serum creatinine as a marker of kidney function: a meta-analysis. Am J Kidney Dis 40:221–226
Meersch M, Schmidt C, Van Aken H, Martens S, Rossaint J, Singbartl K, Görlich D, Kellum JA, Zarbock A (2014) Urinary TIMP-2 and IGFBP7 as early biomarkers of acute kidney injury and renal recovery following cardiac surgery. PLoS One 9:e93460
Meersch M, Schmidt C, Van Aken H, Rossaint J, Görlich D, Stege D, Malec E, Januszewska K, Zarbock A (2014) Validation of cell-cycle arrest biomarkers for acute kidney injury after pediatric cardiac surgery. PLoS One 9:e110865
Kwiatkowski DM, Goldstein SL, Krawczeski CD (2012) Biomarkers of acute kidney injury in pediatric cardiac patients. Biomarkers 6:273–282
Chawla LS, Kellum JA (2012) Acute kidney injury in 2011: biomarkers are transforming our understanding of AKI. Nat Rev Nephrol 8:68–70
Vanmassenhove J, Vanholder R, Nagler E, Van Biesen W (2013) Urinary and serum biomarkers for the diagnosis of acute kidney injury: an in-depth review of the literature. Nephrol Dial Transplant 28:254–273
Basu RK, Wong HR, Krawczeski CD, Wheeler DS, Manning PB, Chawla LS, Devarajan P, Goldstein SL (2014) Combining functional and tubular damage biomarkers improves diagnostic precision for acute kidney injury after cardiac surgery. J Am Coll Cardiol 64:2753–2762
Abuelo JG (2007) Normotensive ischemic acute renal failure. N Engl J Med 357:797–805
Devarajan P (2006) Update on mechanisms of ischemic acute kidney injury. J Am Soc Nephrol 17:1503–1520
Rosner MH, Okusa MD (2006) Acute kidney injury associated with cardiac surgery. Clin J Am Soc Nephrol 1:19–32
Okusa MD (2002) The inflammatory cascade in acute ischemic renal failure. Nephron 90:133–138
Algaze CA, Koth AM, Faberowski LW, Hanley FL, Krawczeski CD, Axelrod DM (2017) Acute kidney injury in patients undergoing the extracardiac Fontan operation with and without the use of cardiopulmonary bypass. Pediatr Crit Care Med 18:34–43
Stafford-Smith M, Podgoreanu M, Swaminathan M, Phillips-Bute B, Mathew JP, Hauser EH, Winn MP, Milano C, Nielsen DM, Smith M (2005) Association of genetic polymorphisms with risk of renal injury after coronary bypass graft surgery. Am J Kidney Dis 45:519–530
Cardinal-Fernández P, Ferruelo A, Martín-Pellicer A, Nin N, Esteban A, Lorente J (2012) Genetic determinants of acute renal damage risk and prognosis: a systematic review. Med Intensiva (English Edition) 36:626–633
Lassnigg A, Schmid ER, Hiesmayr M, Falk C, Druml W, Bauer P, Schmidlin D (2008) Impact of minimal increases in serum creatinine on outcome in patients after cardiothoracic surgery: do we have to revise current definitions of acute renal failure? Crit Care Med 36:1129–1137
Cooper DS, Claes D, Goldstein SL, Menon S, Bennett M, Ma Q, Krawczeski C (2013) Novel urinary biomarkers remain elevated years after acute kidney injury following cardiac surgery in children. J Am Coll Cardiol 61:E438
Nichols DG, Greeley WJ, Lappe DG, Ungerleider RM, Cameron DE, Spevak PJ, Wetzel RC (2006) Critical heart disease in infants and children. Elsevier Health Science, Amsterdam, pp 113–130
Sutherland SM, Zappitelli M, Alexander SR, Chua AN, Brophy PD, Bunchman TE, Hackbarth R, Somers MJ, Baum M, Symons JM, Flores FX, Benfield M, Askenazi D, Chand D, Fortenberry JD, Mahan JD, McBryde K, Blowey D, Goldstein SL (2010) Fluid overload and mortality in children receiving continuous renal replacement therapy: the prospective pediatric continuous renal replacement therapy registry. Am J Kidney Dis 55:316–325
Hassinger AB, Wald EL, Goodman DM (2014) Early postoperative fluid overload precedes acute kidney injury and is associated with higher morbidity in pediatric cardiac surgery patients. Pediatr Crit Care Med 15:131–138
Wilder NS, Yu S, Donohue JE, Goldberg CS, Blatt NB (2016) Fluid overload is associated with late poor outcomes in neonates following cardiac surgery. Pediatr Crit Care Med 17:420–427
Costello JM, Thiagarajan RR, Dionne RE, Allan CK, Booth KL, Burmester M, Wessel DL, Laussen PC (2006) Initial experience with fenoldopam after cardiac surgery in neonates with an insufficient response to conventional diuretics. Pediatr Crit Care Med 7:28–33
Axelrod DM, Anglemyer AT, Sherman-Levine SF, Zhu A, Grimm PC, Roth SJ, Sutherland SM (2014) Initial experience using aminophylline to improve renal dysfunction in the pediatric cardiovascular ICU. Pediatr Crit Care Med 15:21–27
Axelrod DM, Sutherland SM, Anglemyer A, Grimm PC, Roth SJ (2016) A double-blinded, randomized, placebo-controlled clinical trial of aminophylline to prevent acute kidney injury in children following congenital heart surgery with cardiopulmonary bypass. Pediatr Crit Care Med 17:135–143
Ricci Z, Stazi GV, Di Chiara L, Morelli S, Vitale V, Giorni C, Ronco C, Picardo S (2008) Fenoldopam in newborn patients undergoing cardiopulmonary bypass: controlled clinical trial. Interact Cardiovasc Thorac Surg 7:1049–1053
Costello JM, Masterson CD, Allan CK, Gauvreau K, Newburger JW, McGowan FX, Wessel DL, Mayer JE, Salvin JW, Dionne RE (2014) Impact of empiric nesiritide or milrinone infusion on early postoperative recovery following Fontan surgery: a randomized, double-blind, placebo-controlled trial. Circ Heart Fail 7:596–604
Sampath S, Moran JL, Graham PL, Rockliff S, Bersten AD, Abrams KR (2007) The efficacy of loop diuretics in acute renal failure: assessment using Bayesian evidence synthesis techniques. Crit Care Med 35:2516–2524
Mehta RL, Pascual MT, Soroko S, Chertow GM, Group PS (2002) Diuretics, mortality, and nonrecovery of renal function in acute renal failure. JAMA 288:2547–2553
Van der Vorst MM, Kist JE, van der Heijden AJ, Burggraaf J (2006) Diuretics in pediatrics. Pediatr Drugs 8:245–264
Madenci AL, Stoffan AP, Rajagopal SK, Blinder JJ, Emani SM, Thiagarajan RR, Weldon CB (2013) Factors associated with survival in patients who undergo peritoneal dialysis catheter placement following cardiac surgery. J Pediatr Surg 48:1269–1276
Madenci AL, Thiagarajan RR, Stoffan AP, Emani SM, Rajagopal SK, Weldon CB (2013) Characterizing peritoneal dialysis catheter use in pediatric patients after cardiac surgery. J Thorac Cardiovasc Surg 146:334–338
Bojan M, Gioanni S, Vouhé PR, Journois D, Pouard P (2012) Early initiation of peritoneal dialysis in neonates and infants with acute kidney injury following cardiac surgery is associated with a significant decrease in mortality. Kidney Int 82:474–481
Kwiatkowski DM, Menon S, Krawczeski CD, Goldstein SL, Morales DL, Phillips A, Manning PB, Eghtesady P, Wang Y, Nelson DP (2015) Improved outcomes with peritoneal dialysis catheter placement after cardiopulmonary bypass in infants. J Thorac Cardiovasc Surg 149:230–236
Sasser WC, Dabal RJ, Askenazi DJ, Borasino S, Moellinger AB, Kirklin JK, Alten JA (2014) Prophylactic peritoneal dialysis following cardiopulmonary bypass in children is associated with decreased inflammation and improved clinical outcomes. Congenit Heart Dis 9:106–115
Goldstein SL, Somers MJ, Baum MA, Symons JM, Brophy PD, Blowey D, Bunchman TE, Baker C, Mottes T, Mcafee N (2005) Pediatric patients with multi-organ dysfunction syndrome receiving continuous renal replacement therapy. Kidney Int 67:653–658
Stromberg D, Fraser CD Jr, Sorof JM, Drescher K, Feltes TF (1997) Peritoneal dialysis. An adjunct to pediatric postcardiotomy fluid management. Tex Heart Inst J 24:269
Alkan T, Akçevin A, Türkoglu H, Paker T, Sasmazel A, Bayer V, Ersoy C, Askn D, Aytaç A (2006) Postoperative prophylactic peritoneal dialysis in neonates and infants after complex congenital cardiac surgery. ASAIO J 52:693–697
Chien J-C, Hwang B-T, Weng Z-C, Meng LC-C, Lee P-C (2009) Peritoneal dialysis in infants and children after open heart surgery. Pediatr Neonatol 50:275–279
Averbuch N, Birk E, Frenkel G, Gogia O, Shulman OM, Bruckheimer E, Nachum E, Amir G (2014) Percutaneous intraperitoneal catheters in neonates following open heart surgery. J Intensive Care Med 29:160–164
Kwiatkowski DM, Goldstein SL, Cooper DS, Nelson DP, Morales DS, Krawczeski CD (2017) Peritoneal dialysis vs furosemide for prevention of fluid overload in infants after cardiac surgery: a randomized clinical trial. JAMA Pediatr. doi:10.1001/jamapediatrics.2016.4538
Riley AA, Jefferies JL, Nelson DP, Bennett MR, Blinder JJ, Ma Q, Devarajan P, Goldstein SL (2014) Peritoneal dialysis does not adversely affect kidney function recovery after congenital heart surgery. Int J Artif Organs 37:39–47
Fleming GM, Askenazi DJ, Bridges BC, Cooper DS, Paden ML, Selewski DT, Zappitelli M (2012) A multicenter international survey of renal supportive therapy during ECMO: the Kidney Intervention During Extracorporeal Membrane Oxygenation (KIDMO) group. ASAIO J 58:407–414
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflicts of interest.
Additional information
Key summary points
1. AKI is common in infants and children after cardiac surgery with CPB.
2. In this cohort, AKI is associated with worse outcomes, including mortality.
3. Diagnosis of AKI is impaired by the reliance on serum creatinine.
4. Fluid overload is one of the major determinants of morbidity and mortality in children with postoperative AKI.
5. Early peritoneal dialysis is a safe and effective method of fluid removal in patients after congenital heart surgery.
Answers
1. d; 2. a; 3. d; 4. e; 5. b
Questions (answers are provided following the reference list)
Questions (answers are provided following the reference list)
-
1.
Using KDIGO AKI criteria, what minimum creatinine change defines AKI?
-
a)
Creatinine increase of 0.3 mg/dl
-
b)
Creatinine increase of 0.5 mg/dl
-
c)
Creatinine increase of 2 times baseline
-
d)
Creatinine increase of 0.3 mg/dl or 1.5 times baseline
-
e)
Creatinine increase of 0.5 mg/dl or 2 times baseline
-
a)
-
2.
What is the most common timeframe for AKI after CPB in infants and children?
-
a)
First 24–48 h, lasting for 1–2 days
-
b)
First 24–48 h, lasting for 4 days
-
c)
First 72–96 h, lasting for 1–2 days
-
d)
First 72–96 h, lasting for 4 days
-
e)
Greater than 7 days, lasting for 1 day
-
a)
-
3.
Which of the following is a biomarker of renal tubule structural injury?
-
a)
Cystatin-C
-
b)
Creatinine
-
c)
Urine output
-
d)
NephroCheck
-
a)
-
4.
Which of the following is not associated with fluid overload?
-
a)
Decreased nutritional absorption
-
b)
Increased rate of infection
-
c)
Decreased myocardial contraction
-
d)
Increased mortality
-
e)
Increased lung compliance
-
a)
-
5.
In an infant with oliguria after cardiac surgery, which of the following therapies is shown to be associated with less fluid overload and a lower rate of prolonged mechanical ventilation?
-
a)
Dobutamine
-
b)
Peritoneal dialysis
-
c)
Nesiritide
-
d)
Furosemide
-
e)
Aminophylline
-
a)
Rights and permissions
About this article

Cite this article
Kwiatkowski, D.M., Krawczeski, C.D. Acute kidney injury and fluid overload in infants and children after cardiac surgery. Pediatr Nephrol 32, 1509–1517 (2017). https://doi.org/10.1007/s00467-017-3643-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-017-3643-2