
Abstract
Background
Eculizumab may be used to treat C3-glomerulopathy (C3G), a rare but severe glomerular disease.
Diagnosis and Treatment
Patients 1, 2 and 3 were diagnosed with nephritic syndrome with alternative complement pathway activation (low C3, C3Nef-positive) and C3G at the age of 9, 13 and 12 years, respectively. Treatment with eculizumab normalized proteinuria within 1, 2 and 7 months, respectively. Proteinuria relapsed when eculizumab was withdrawn, but the re-introduction of eculizumab normalized proteinuria. Patient 4 was diagnosed with C3G at 9 years of age, with progression to end-stage renal disease within 2 years, followed by a first renal transplantation (R-Tx) with early disease recurrence and graft loss within 39 months. After a second R-Tx, she rapidly presented with biological and histological recurrence: therapy with eculizumab was started, with no effect on proteinuria after 5 months, in a complex clinical setting (i.e. association of C3G recurrence, humoral rejection and BK nephritis). Eculizumab was withdrawn due to multiple viral reactivations, but the re-introduction of the drug a few months later enabled a moderate decrease in proteinuria.
Conclusion
These cases illustrate the efficacy of eculizumab, at least on native kidneys, in paediatric C3G. However, larger international studies are warranted to confirm the benefit and safety of eculizumab therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary membranoproliferative glomerulonephritis (MPGN) was traditionally classified into type I (characterized by subendothelial deposits), type II (dense deposits in the glomerular membrane basement) and type III (both subepithelial and subendothelial deposits) according to histopathological pattern [1]. Advances in the understanding of the disease have recently led to a new classification, based on the pathogenesis of the disease process. Indeed, this classification introduces the concept of MPGN related to immunoglobulin-mediated disease (i.e. caused by classical complement pathway activation) in contrast to non-immunoglobulin-mediated disease (i.e. caused by alternative complement pathway activation). As such, new subgroups have been proposed, namely immunoglobulin (Ig)-associated MPGN, MPGN with dominant C3 [the ‘C3 glomerulopathies’ (C3G)] and idiopathic MPGN [2]. This newly acquired understanding of the pathogenesis of C3G offers novel therapeutic opportunities to target complement pathways.
The C5 blocker eculizumab is a humanized monoclonal antibody directed against human complement component C5 that blocks the activation of the alternative complement pathway. Its use has dramatically increased in nephrology in recent years. Eculizumab was initially developed for paroxysmal nocturnal hemoglobinuria [3], but it is now widely used for atypical haemolytic uraemic syndrome [4] and also as ‘off label’ therapy for C3G, humoral rejection and typical haemolytic uraemic syndrome. Results are nevertheless conflicting and are usually from small series and reports, at least in paediatric nephrology [5].
The incidence of C3G is estimated to be 1–2 per 106 children, occurring essentially between 5 and 15 years of age. The clinical presentation can consist of nephrotic syndrome (20% of cases), nephritic syndrome (20% of cases), asymptomatic haematuria or proteinuria (60% of cases), but also isolated hypertension, or discovery during an assessment of renal failure [6]. C3G accounts for 2% of the primary diagnoses in children who receive renal transplantation (R-Tx). After R-Tx, the overall rate of recurrence has been reported between 30 and 77%, with graft failure due to recurrence in 17–50% of recipients. Time to recurrence ranges between 0.5 and 2 years, with the first sign being a late-onset proteinuria [7]. The risk of recurrence and graft loss is independent of initial clinical presentation and C3 concentration, but is strongly associated with the presence of proteinuria, which is the hallmark of clinical recurrence. Van Stralen et al. reported that paediatric patients receiving R-Tx for C3G presented an increased risk of graft loss at 5 years in comparison to patients with uropathies [8]. These authors additionally found that patients with C3G also displayed an ongoing high risk of graft loss over time [8].
Here we report a single-center experience in managing four paediatric patients with C3G who received eculizumab, of whom three received eculizumab for C3G of their native kidneys and the fourth because of early recurrence after a second R-Tx.
Cases
Patients 1 and 2 are brother (Pt1, age 9 years ) and sister (Pt2, age 13 years), born to non-consanguineous parents, who presented with persistent proteinuria for several months, microscopic hematuria, normal renal function and alternative complement pathway activation (very low C3 and CH50 levels, positive C3nef). Renal biopsy showed C3G with C3 deposits of similar severity in the two siblings, although the clinical phenotype was more severe in Pt2 (nephrotic proteinuria). No mutations of the complement pathway were found (VFB). The initial management of Pt2 included treatment with corticosteroids (60 mg/m2/day) and angiotensin-converting enzyme (ACE) inhibitors for 2 months, followed by the addition of mycophenolate mofetil (MMF, 1500 mg/day) and an increase in ACE inhibitors for 1 month. However, all these measures remained ineffective. Therapy with eculizumab was commenced in Pt1 and Pt2 in the absence of other therapies except for ACE inhibitors and angiotensin II receptor antagonists, eculizumab (900 mg every 2 weeks) normalized proteinuria within 1 and 2 months, respectively. However, even though clinical evolution was successful, control biopsies (performed after 18 and 9 months of eculizumab in Pt1 and Pt2, respectively) found neither improvement nor progression of the disease, without any modifications in C3 staining. With these results in mind, due to financial and practical constraints, it was decided to withdraw eculizumab therapy in both siblings after a French multidisciplinary review of the cases. However, a rapid biological relapse was observed in the two children, which necessitated that the eculizumab therapy be restarted. The re-introduction of the drug led to a rapid decrease of proteinuria, as illustrated in Fig. 1a, b. Notably, Pt2 presented with invasive Neisseria meningitidis Y 3 months after restarting eculizumab therapy, despite antibiotic prophylaxis with phenoxymethylpenicilline (but likely non-satisfactory compliance) and anti-Neisseria meningitis immunizations as recommended by international guidelines. She recovered from this acute infectious complication without any sequelae.

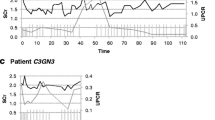
Evolution of urinary protein/creatinine ratio in patient 1 (a; 9-year old boy), patient 2 (b; 12-year old girl), patient 3 (c; 12-year old boy) and patient 4 (d; 17-year-old girl) according to time and eculizumab therapy. ECZ Eculizumab, red arrows start and stop of eculizumab therapy, black arrows plasma exchange, black stars intravenous immunoglobulins
Patient 3 (Pt3) is a 12-year-old boy who presented with nephrotic proteinuria, gross hematuria and renal failure (maximum serum creatinine of 176 μmol/L); C3 levels were decreased while C4 levels were normal, and C3nef was present. Genetic analysis was not performed. Renal biopsy showed C3G with IgA, IgG and IgM, C3 and C1q deposits, leading us to diagnose C3G without completely ruling out an idiopathic or immune-mediated glomerulopathy. Systemic lupus was also ruled out (absence of associated clinical symptoms, negative biological investigations). The patient was started on ‘conventional’ therapy of corticosteroids, MMF, ACE inhibitors and angiotensin II receptor antagonists, with the addition of eculizumab (900 mg every 2 weeks) after 4 weeks of therapy; renal function was stabilized within 5 months, and the urinary protein/creatinine ratio progressively decreased from 2046 to 30 mg/mmol after 7 months of therapy. We attempted to withdraw eculizumab after 8 months of treatment without performing a renal biopsy, but the patient relapsed within 1 month, thus requiring restarting of the eculizumab therapy. A complete clinical and biological recovery was observed 2 months after treatment with eculizumab was restarted, as illustrated in Fig. 1c.
Patient 4 (Pt4) is a 17-year-old girl with C3G diagnosed abroad at 9 years of age, with progression to end-stage renal disease within 2 years and a first R-Tx with early recurrence and graft loss within 39 months. Study of the alternative complement pathway did not reveal any abnormality, and C3nef was absent. She was referred to our center for a second R-Tx, after which she rapidly presented with biological and histological recurrence (biopsy performed 6 weeks after R-Tx). She was then started on eculizumab, with no effect on proteinuria after 5 months. A second biopsy performed 8 months after R-Tx revealed a complex combination of recurrence, humoral rejection and BK nephritis. Eculizumab therapy was withdrawn temporarily due to multiple viral reactivations in order to avoid infectious complications. In addition to immunosuppressive therapies (induction with thymoglobulins, corticosteroids, MMF and tacrolimus), she also received plasma exchanges and monthly intravenous immunoglobulins. Thirteen months after R-Tx, the viral reactivations were controlled, allowing us to reintroduce eculizumab (900 mg every 2 weeks): after 8 weeks of eculizumab, without any significant changes in other therapies, the urinary protein/creatinine ratio decreased from 364 to 70 mg/mmol (Fig. 1d).
Pt1 and Pt2 displayed increased C5b9 levels (fourfold the upper normal limit) during the acute phase of C3G, whereas Pt3 and Pt4 had normal C5b9 levels. In all patients, low circulating C3 levels were observed during the entire follow-up.
The clinical, biological and quantitative pathological data on these patients are summarized in Table 1.
Discussion
The four cases presented here illustrate the efficacy and safety of eculizumab for treating paediatric C3G, with three patients receiving eculizumab therapy on native kidneys and the last one receiving eculizumab because of early recurrence after a second R-Tx. The three patients treated on their native kidneys achieved complete clinical and biological remission. Due to the rarity and the severity of the disease, our report of eculizumab therapy for paediatric C3G in our center is exhaustive. Fewer than 20 cases of paediatric C3G have been reported to date, and the clinical presentations have been heterogeneous, and conventional immunosuppressive therapies (such as MMF) have not been uniformly useful [9, 10].
In this series, we describe the absence of histological response in the two patients in whom we performed a control biopsy, unlike some cases reported to date [11]. To explain such discrepancies, we can hypothesize that we performed the control biopsy too early after eculizumab initiation (i.e. after 9 and 18 months of therapy), but it does not seem ethical to propose a new biopsy to these patients now that their disease is under control. It would have been interesting to have the results of an electron microscopy analysis but unfortunately it was not performed. In Pt3, a second biopsy could have shown a more clear-cut immunofluorescence, leading to a definitive diagnosis. We have to acknowledge one serious side effect of eculizumab therapy, i.e. invasive infection to Neisseria meningitidis despite meningococcal vaccines and antibiotic prophylaxis with phenoxymethylpenicillin; these measures were performed according to international guidelines, however in a context of non-optimal compliance. It is therefore important to warn patients and families of this possible life-threatening complication, but we believe that the pros and cons of eculizumab therapy in paediatric C3G favors the use of such a drug. Even though there is no specific rationale to withdraw eculizumab in case of viral infections, we decided to stop it in Pt4 since we believed that we should use it with caution in case of systemic infection (whatever the exact etiology), especially in a context of ‘off-label’ use.
One main limitation of this report is that we cannot completely rule out a spontaneous remission of C3G due to the possible natural evolution of C3G. However, we consider it very unlikely that a spontaneous remission due to a natural cause would occur in all our patients after having received eculizumab. Pt1 had low proteinuria before starting eculizumab, which clouds interpretation of his initial response to this drug, but at the end of treatment his proteinuria had relapsed to high levels. Finally, we cannot rule out a beneficial effect of the other drugs used concomitantly in our patients (especially in Pt3), namely immunosuppressive therapies (mainly MMF) and ACE inhibitors, as already mentioned in previous reports [12, 13]. However, a number of other authors have already discussed the growing interest in using eculizumab in paediatric refractory C3G [14]. In our first three patients, the timing of eculizumab withdrawal/end of clinical and biological follow-up/early relapse/rapid re-introduction of eculizumab and eventually complete remission without any significant changes in the other therapies provides a strong rationale for a beneficial effect of eculizumab per se on C3G, as illustrated in Fig. 1.
Based on our limited experience of paediatric C3G on native kidneys, we therefore believe that some key findings can be highlighted, however they should be interpreted with caution due to the limited number of patients. First, eculizumab can induce clinical and biological response, but not necessarily histological response. Second it is necessary to wait several months before drawing any conclusion on the effectiveness (or not) of eculizumab therapy in paediatric C3G. Three, it can be useful to withdraw eculizumab even in case of complete remission, with a close follow-up and a rapid re-introduction of eculizumab in case of relapse. Fourth, conventional immunosuppression should be attempted in patients with C3G before therapy with eculizumab is considered; however, should treatment with eculizumab be started, maintenance immunosuppressive adjuvant therapies (MMF) need to be re-evaluated. Fifth, patients and families should be aware of possible infectious complications, requiring a good compliance to antibiotic prophylaxis.
In contrast, for the patient receiving eculizumab after R-Tx, it is impossible to draw conclusions from this case report because on the one hand the clinical picture was complex (i.e. association of C3G recurrence, humoral rejection and BK nephritis), and on the other hand the patient received many other therapies, including plasma exchange, that could have had beneficial effects on C3G and on the other comorbidities. Moreover, eculizumab therapy probably helped to control proteinuria, but it is impossible to distinguish between the effect of the drug on the recurrence of C3G and/or the effect on humoral rejection [15].
Conclusion
These four cases illustrate that eculizumab therapy is an efficient but expensive treatment of paediatric C3G, especially in patients with native kidneys. The total duration of treatment remains undetermined in the absence of long-term data, both in our study and in the literature. Larger international studies are therefore urgently required to confirm the benefits and safety of eculizumab in this indication.
References
Sethi S, Fervenza FC (2012) Membranoproliferative glomerulonephritis—a new look at an old entity. N Engl J Med 366:1119–1131
Bomback AS, Appel GB (2012) Pathogenesis of the C3 glomerulopathies and reclassification of MPGN. Nat Rev Nephrol 8:634–642
Bauters T, Bordon V, Robays H, Benoit Y, Dhooge C (2012) Successful use of eculizumab in a pediatric patient treated for paroxysmal nocturnal hemoglobinuria. J Pediatr Hematol Oncol 34:e346–e348
Fakhouri F, Delmas Y, Provot F, Barbet C, Karras A, Makdassi R, Courivaud C, Rifard K, Servais A, Allard C, Besson V, Cousin M, Châtelet V, Goujon JM, Coindre JP, Laurent G, Loirat C, Frémeaux-Bacchi V (2014) Insights from the use in clinical practice of eculizumab in adult patients with atypical hemolytic uremic syndrome affecting the native kidneys: an analysis of 19 cases. Am J Kidney Dis 63:40–48
Bomback AS (2014) Eculizumab in the treatment of membranoproliferative glomerulonephritis. Nephron Clin Pract 128:270–276
Servais A, Noël LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, Macher MA, Zuber J, Karras A, Provot F, Moulin B, Grünfeld JP, Niaudet P, Lesavre P, Frémeaux-Bacchi V (2012) Acquired and genetic complement abdnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int 82:454–464
Bacchetta J, Cochat P (2015) Primary disease recurrence-effects on paediatric renal transplantation outcomes. Nat Rev Nephrol 11:371–384
Van Stralen KJ, Verrina E, Belingheri M, Dudley J, Dusek J, Grenda R, Macher MA, Puretic Z, Rubic J, Rudaitis S, Rudin C, Schaefer F, Jager KJ (2013) Impact of graft loss among kidney diseases with a high risk of post-transplant recurrence in the paediatric population. Nephrol Dial Transplant 28:1031–1038
Nicolas C, Vuiblet V, Baudouin V, Macher MA, Vrillon I, Biebuyck-Gouge N, Dehennault M, Gie S, Morin D, Nivet H, Nobili F, Ulinski T, Ranchin B, Marinozzi MC, Ngo S, Fremeaux-Bacchi V, Pietrement C (2014) C3 nephritic factor associated with C3 glomerulopathy in children. Pediatr Nephrol 29:85–94
Okuda Y, Ishikura K, Hamada R, Harada R, Sakai T, Hamasaki Y, Hataya H, Fukuzawa R, Ogata K, Honda M (2015) Membranoproliferative glomerulonephritis and C3 glomerulonephritis : frequency, clinical features, and outcome in children. Nephrology 20:286–292
Vivarelli M, Pasini A, Emma F (2012) ECULIZUMAB for the treatment of dense deposit disease. N Engl J Med 366:1163–1165
De S, Al-Nabhani D, Thorner P, Cattran D, Piscione TD, Licht C (2009) Remission of resistant MPGN type I with mycophenolate mofetil and steroids. Pediatr Nephrol 24:597–600
Rabasco C, Cavero T, Román E, Rojas-Rivera J, Olea T, Espinosa M, Cabello V, Fernández-Juarez G, González F, Ávila A, Baltar JM, Díaz M, Alegre R, Elías S, Antón M, Frutos MA, Pobes A, Blasco M, Martín F, Bernis C, Macías M, Barroso S, de Lorenzo A, Ariceta G, López-Mendoza M, Rivas B, López-Revuelta K, Campistol JM, Mendizábal S, de Córdoba SR, Praga M, Spanish Group for the Study of Glomerular Diseases (GLOSEN) (2015) Effectiveness of mycophenolate mofetil in C3 glomerulonephritis. Kidney Int 88:1153–1160
Rousset-Rouvière C, Cailliez M, Garaix F, Bruno D, Laurent D, Tsimaratos M (2014) Rituximab fails where eculizumab restores renal function in C3nef related DDD. Pediatr Nephrol 29:1107–1111
González-Roncero F, Suñer M, Bernal G, Cabello V, Toro M, Pereira P, Angel Gentil M (2012) Eculizumab treatment of acute antibody-mediated rejection in renal transplantation: case reports. Transplant Proc 44:2690–2694
Acknowledgements
The authors would like to thank Dr Marie-Nathalie Sarda (Department of Biology, Hospices Civils de Lyon) for her help in local complement assessment, and also Dr Moglie Le Quintrec, Pr Eric Rondeau, Pr Chantal Loirat, Pr Fadi Fakhouri and Dr Sophie Chauvet for their expertise during the French multidisciplinary review of the first two cases.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics
The local ethical committee (CPP Lyon Sud Est II) approved this retrospective review of cases (IRB 00009118, session 1/27/2016).
Disclosure of interest
JB received travel grants from Alexion. ALL received travel grants from Alexion and honararia from Alexion (2013, 2014). VFB acts as a scientific advisor for Alexion.
Rights and permissions
About this article

Cite this article
Lebreton, C., Bacchetta, J., Dijoud, F. et al. C3 glomerulopathy and eculizumab: a report on four paediatric cases. Pediatr Nephrol 32, 1023–1028 (2017). https://doi.org/10.1007/s00467-017-3619-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-017-3619-2