
Abstract
Background
Hyperactivity of the alternative complement pathway is the principle defect in C3 glomerulopathies (C3G). Eculizumab, a monoclonal antibody that binds C5 to prevent formation of the membrane attack complex, has been shown to be beneficial in some patients with this disease.
Methods
In this open-label, proof-of-concept efficacy-and-safety study, a patient with the initial diagnosis of dense deposit disease (DDD) and allograft recurrence of C3 glomerulonephritis (C3GN) was treated with eculizumab every other week for 1 year. The patient had pathological evidence of C3GN and proteinuria >1 g/day at enrollment. He underwent graft biopsy before enrollment and repeat biopsy at 6 and 12 months.
Results
Although no mutations were identified in complement genes, functional studies were positive for C3 nephritic factors and elevated levels of soluble membrane attack complex (sMAC). On therapy, sMAC levels normalized and although proteinuria initially decreased, it increased reaching pre-treatment levels at 12 months. Although serum creatinine remained stable, repeat allograft biopsies showed progression of disease.
Conclusions
Clinical and histopathologic data suggest a partial response to eculizumab in this patient. While eculizumab blocked activation of the terminal complement cascade, persistent dysregulation of the alternative pathway remained, indicating eculizumab alone cannot control disease in this patient. Additional research is required to identify effective anticomplement therapy for this group of C3G patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Membranoproliferative glomerulonephritis (MPGN) denotes a general pattern of glomerular injury characterized by an increase in mesangial cellularity and matrix with thickening of glomerular capillary walls secondary to subendothelial deposition of immune complexes and/or complement factors, cellular entrapment, and new basement membrane formation. This pattern of injury is easily noted by light microscopy, making the diagnosis of MPGN relatively straightforward; however, immunofluorescence (IF) and electron microscopy (EM) reveal differences within MPGN and have been used to define MPGN subgroups.
Subgrouping is driven under the presumption that histological differences are reflective of underlying pathogenesis, which may affect clinical care. Based on EM description of electron-dense deposits relative to the glomerular basement membrane (GBM), such as subendothelial, intramembranous (within the lamina densa), or both subendothelial and subepithelial, MPGN is classified as type I, II, or III, respectively [1, 2]. Immunofluorescence studies typically reveal immunoglobulins (usually IgG or IgM) in MPGN type I and type III, but not in type II. Although the three MPGN types stain positive for complement component 3 (C3) consistent with complement activation, C3-positive but Ig-negative examples of MPGN type I and MPGN type III exist [3]. Together with MPGN type II, more appropriately called dense deposit disease (DDD), this group of C3-positive Ig-negative glomerular diseases has recently been named C3 glomerulopathies (C3G) [4]. Two broad subtypes of C3G are recognized: DDD, characterized by amorphous, extremely dense, sausage-shaped intramembranous GBM EM deposits; and C3GN, characterized by subendothelial, occasional subepithelial and mesangial glomerular EM deposits that have more lobular, fuzzier, and lighter appearance as compared with the darker and sharper deposits of DDD.
Common to all C3Gs is unrestrained complement activity, which can be caused by mutations in complement genes [e.g., C3 (complement component 3), CFB (factor B), CFH (factor H), CFI (factor I)] or acquired autoantibodies that stabilize C3 convertase, the activating complex of the alternative pathway [e.g., C3 nephritic factors (C3Nefs)] or inhibit complement regulators (e.g., factor H autoantibodies, FHAA) [5, 6]. Nonspecific immunomodulatory therapies, such as corticosteroids, cyclophosphamide, and calcineurin inhibitors have been used in small numbers of C3G patients (both DDD and C3GN) with varied results [7]. No complement-specific therapies have been available to treat these diseases until the recent approval by the Food and Drug Administration of eculizumab, a humanized monoclonal antibody that binds with high affinity to C5 thereby blocking the terminal complement complex and preventing the generation of the membrane attack complex (MAC). Eculizumab has been approved for the treatment of paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS). It has also been used in a small number of patients with C3GN and DDD [8–11]. In this report, we describe a C3G patient who presented with DDD and had allograft recurrence of C3GN. He experienced a partial response to eculizumab therapy. We provide a detailed analysis of complement activity with biomarker levels that suggest that this type of response can be predicted.
Materials and methods
Subjects
This proof-of-concept study of eculizumab (Soliris; Alexion Pharmaceuticals, Cheshire, CT, USA) enrolled one adult patient with C3G. All genetic and complement studies were approved by the Institutional Review Board of University of Iowa. The clinical and research activities being reported are consistent with the principles of the Declaration of Helsinki and with the principles of the Declaration of Istanbul on Organ Trafficking and Transplant Tourism. The patient received meningococcal vaccine prior to initiation of eculizumab therapy and was maintained on oral ciprofloxacin prophylaxis (500 mg daily) during treatment.
Treatment regimen and evaluations
The patient initially received eculizumab at 900 mg intravenously weekly for 4 weeks. On week 5, the dose was increased to 1,200 mg intravenously and was continued at this level every other week for a total treatment period of 53 weeks. This dosing regimen was based on experience using eculizumab for aHUS.
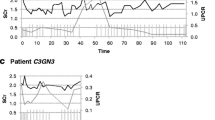
Pharmacokinetic testing was done in the third month of treatment to ensure that dosing regimens were sufficient; no dosing adjustments were needed. During the treatment period, renin angiotensin–aldosterone system–blocking medications were started, based on clinical indications of worsening proteinuria and/or hypertension (Fig. 1). He remained on chronic immunomodulatory therapy with prednisone and tacrolimus for rejection prophylaxis.

Patient’s clinical course after eculizumab therapy. Two months after his second transplant, the patient’s first allograft graft biopsy confirmed recurrent C3GN. Six months after his second transplant, proteinuria prompted the initiation of rituximab therapy. Due to poor response to rituximab, 9 months after his second transplant, eculizumab therapy was started (shown as baseline), which was continued up to 12 months. The patient had repeat biopsies at 6 and 12 months after the initiation of eculizumab therapy (see Fig. 2). Due to persistent urine protein/creatinine ratio (UPr) >1 mg/mg, angiotensin-converting-enzyme inhibitor (ACEi) treatment was started at 9 months
Laboratory measurements performed every 2 weeks during the study period included a basic metabolic panel, complete blood count, hepatic function panel, and spot urine protein-to-creatinine ratio. Blood and urine samples were collected immediately before eculizumab infusions. Baseline screening for mutations in several complement genes (e.g., CFH, CFI, CFHR5, and MCP) and for autoantibodies associated with dysregulation of the alternative pathway (e.g., C3Nefs and FHAA) was performed at the University of Iowa as described previously [12, 13]. In addition, serum and plasma were collected every 4 weeks during the treatment period for functional testing of the complement pathway [12–14].
Outcomes
The primary endpoint was change in proteinuria over the treatment period; secondary endpoints were changes in renal histopathology on biopsies performed at the 6-month and 12-month time points. We also examined trends in serum creatinine and albumin, and changes in alternative complement pathway activity before and during therapy.
Statistical analyses
Given the design of this study, no formal statistical analysis was performed. All data are presented descriptively.
Results
The index patient was a 21-year-old white male with allograft recurrence of C3GN. At the age of 8 years, he was diagnosed with DDD and at age 17, he received his first kidney transplant from a deceased donor (Fig. 2a). A year after his transplant, he developed nephrotic-range proteinuria and a biopsy revealed recurrent disease (Fig. 2b). The patient’s family history was non-contributory and specifically negative for renal diseases. Genetic testing performed by bidirectional sequencing of the coding regions and intron/exon boundaries of CFI, MCP, CFB, C3, and CFHR5 revealed no pathogenic variants although there were three DDD-associated allele variants in CFH. No deletions or duplications were identified over the CFHR1-CFHR4 region. Functional complement studies identified a decrease in the level of alternative pathway serum proteins and an elevation in soluble membrane attack complex (sMAC). There were no autoantibodies to FH but patient-purified immunoglobulins were positive for C3Nefs that stabilized the C3 convertase in assays with and without properdin (C3CSAP and C3CSA, respectively). Consistent with dysregulation of the C3 convertase, C3 breakdown products were identified by immunofixation electrophoresis (IFE) (Table 1).

Renal histopathology in native and graft biopsies. a Original biopsy with mild mesangial cellularity and slight ribbon-like PAS prominence of Bowman’s capsule (arrow), PAS 200× (left panel). Immunofluorescence findings (not illustrated) showed strong C3 staining along glomerular capillary, Bowman’s capsule, and tubular basement membranes. Ultrastructural analysis of the tissue revealed extremely dark, osmophilic, ribbon-like deposits along the glomerular capillary wall within the lamina densa. Similar deposits were seen along the tubular basement membranes and Bowman’s capsule. These findings are diagnostic for DDD (EM×2500) (upper right panel). b H&E of recurrent disease in first allograft. There is mesangial and endocapillary hypercellularity with accentuation of the glomerular lobulations. Many of the infiltrating cells were polymorphonuclear leukocytes (H&E ×200) (left panel). Immunofluorescence (not shown) revealed strong C3 staining along the glomerular capillary wall without associated immunoglobulin. Ultrastructural analysis revealed marked endocapillary inflammation and rare, sausage-shaped, moderately osmophilic deposits within the mesangium (arrow) (EM ×13,500) (right panel). c First biopsy of second transplant demonstrating mesangial and endocapillary proliferation with patchy scarring (PAS 200×) (left panel). There was strong C3 staining along the glomerular capillary wall and in the mesangium (immunofluorescence × 200) (middle panel). Ultrastructural analysis revealed scattered, elongated deposits (arrow), predominantly subendothelial, with endocapillary inflammation and diffuse effacement of the foot processes (EM × 14,800) (right panel). d Comparison of the first (1, 3, 5) and second (2, 4, 6) biopsies in the second allograft revealed slightly greater leukocyte infiltration in the second biopsy and increased chronicity with 20 % fibrosis (2) with similar degrees of mesangial and endocapillary proliferation and slightly more glomeruli demonstrating MPGN features (3, first, 4, second), and similar degrees of tubular atrophy (5 and 6) (1 and 2 H&E 200×; 3 silver 400×; 4 silver 200×; 5 and 6 H&E 100×)
Two years after his initial transplant, the patient received a preemptive second kidney transplant from a living related donor for worsening proteinuria and disease progression. Two months after his transplant, a graft biopsy to monitor disease activity revealed recurrent disease (Fig. 2c). During this time period, serum C3 levels were undetectable and C3Nefs remained positive although the patient was clinically stable with a serum creatinine at 1.5 mg/dl, serum albumin at 4.5 g/dl, and no proteinuria. However within 4 months, the patient started having proteinuria and was placed on rituximab therapy at a fixed dose of 100 mg IV weekly for 4 weeks. This dosing protocol was based on reports of low-dose rituximab efficacy in lymphocyte reduction [15, 16]. Only a minimal clinical and laboratory response was observed, with a 20 % reduction in sMAC levels, stabilization of serum creatinine at 1.5 mg/dl and proteinuria at 2–3 mg/mg. Because sMAC and C3Nefs remained elevated, eculizumab therapy was started.
Eculizumab treatment resulted in complete inhibition of the terminal complement cascade as reflected by normalization of sMAC (<0.30 mg/l on therapy) and was accompanied by a decline in serum creatinine from 1.5 to 1.3 mg/dl (Fig. 1). This improvement in renal function persisted throughout the duration of therapy, with final creatinine in the 1.2–1.4 mg/dl range. The patient’s serum C3 level remained very low during the treatment period (1.27 mg/l pretherapy) although proteinuria initially improved with a random urine protein/creatinine ratio below 0.2 mg/mg after 6 months of eculizumab treatment (Table 2). At 9 months, however, proteinuria increased (persistent urine protein/creatinine ratio >1 mg/mg) and ACEi treatment was initiated (Fig. 1). A sustained and gradual increase in proteinuria necessitated ACEi dose escalation and the addition of angiotensin II receptor blockade (ARB) in an effort to keep the urine protein/creatinine ratio as low as possible. Allograft biopsies at 6 and 12 months of treatment revealed increased chronicity, with 20 % fibrosis in the final biopsy (increased from 3 % in initial biopsy) and continuously active C3GN with persistent membranoproliferative changes and large subendothelial deposits (Fig. 2d). There were no adverse events or infections during the course of therapy and patient remains on bimonthly eculizumab treatment at the time of this report with a stable serum creatinine of 1.2–1.4 mg/dl and urine protein/creatinine ratio of 1.3 mg/mg creatinine on maximum dose of ACEi and ARB therapy.
Discussion
Here we present our experience using rituximab and eculizumab, monoclonal antibodies against CD20 and C5, respectively, for recurrent C3GN. In this proof-of-concept study, we show that eculizumab effectively inhibits the terminal complement cascade and is well tolerated, but only partially prevented progression of C3GN in this patient. Baseline genetic screening coupled with functional assays of the complement pathway support an incomplete response to treatment and is consistent with our understanding of the pathophysiology of C3G (Fig. 3) [17].

The pathophysiology of C3Gs. The C3Gs include DDD and C3GN and share a common dysregulation at the level of the C3 and C5 convertases. It is possible that the relative degree of convertase dysregulation differs between dense deposit disease (DDD) and C3GN. Eculizumab is a monoclonal antibody to C5 that prevents cleavage of C5 to C5a and C5b. In the absence of C5b, membrane attack complex (MAC) and soluble membrane attack complex (sMAC) cannot be generated, thus halting the contribution of the terminal complement cascade to the disease process. However, more proximal dysregulation at the level of the C3 convertase of the alternative pathway continues unchecked, the consequence of which is progression of disease
This response is also consistent with insights provided by three familial studies of DDD and C3GN. One family described by Martínez-Barricarte et al. included a mother and her two identical twin boys, all of whom segregated a 2-amino acid deletion in MG7 of C3 (D923-924AspGly) [18]. This mutation makes the mutant C3 resistant to cleavage by C3 convertase; however, a hydrolyzed mutant C3 convertase does form that cleaves circulating wild-type C3 and resists FH regulation. The functional consequence is persistent fluid-phase mutant C3 convertase activity and the clinical sequela is DDD caused exclusively by fluid-phase alternative pathway dysregulation without any contribution of the terminal complement complex (TCC). In this family, sMAC was not elevated and eculizumab would not be a sensible treatment option.
A second familial study reported by Gale et al. describes two Cypriot families segregating autosomal dominant microscopic hematuria [19]. Renal biopsy was remarkable for C3GN, and a genome-wide linkage study localized a genetic abnormality to the CFH/CFHR region of chromosome 1q31-32. A novel CFHR5 fusion was identified that altered control of both the C3 and C5 convertases in a dominant-negative manner. This finding and mechanism of action are supported by other reports of CFHR5 mutations in patients with DDD and C3GN [12].
In the third family study, reported by Habbig et al., two siblings of consanguineous parentage developed childhood-onset hematuria and proteinuria [20]. Serum C3 and CFB were decreased and C3 breakdown products were increased. Prominent mesangial deposition of C3 and C5b-9 was noted on renal biopsy, and by electron microscopy, there were numerous osmophilic mesangial deposits with intramembranous and subendothelial deposits, consistent with C3GN. Both children were homozygous for the deletion of a lysine at position 224 of CFH (CFHK244), the functional consequence of which was reduced cofactor activity, decay-accelerating activity and C3b binding. In both this family and the Cypriot families, dysregulation of the C5 convertase could be documented and eculizumab would be expected to offer some benefit.
These families and our case underscore an important point: the C3Gs are a disease spectrum in which the level and degree of dysregulation of both the alternative pathway and the TCC vary. Therefore, complement-targeting treatment must be individualized. Today, unfortunately, only TCC targeting is possible, and although eculizumab is extremely effective in preventing the generation of C5a and MAC, it is unlikely to prevent progression of disease if dysregulation occurs higher upstream. In patients with minimal dysregulation of the TCC, the benefit of eculizumab is minimal. The degree of dysregulation of the TCC can be inferred from sMAC testing, which should be obtained in all C3G patients prior to eculizumab therapy [9, 21].
In the patient we have presented, dysregulation of the C3 convertase leads to fluid-phase consumption of C3. We identified C3Nefs (IgG antibodies) that directly stabilize the C3 convertase to prevent its normal physiologic regulation. C3Nefs are detected in approximately 80 % of DDD patients, however their measurement requires specialized testing, which may explain why C3Nefs levels do not always correlate with plasma C3 consumption and disease severity [21–24]. This patient also had dysregulation of the TCC at the level of the C5 convertase as evidenced by elevation of sMAC levels. In an earlier report, we have shown that elevation of sMAC (i.e., sMAC >0.3 mg/l) can be used to predict response to eculizumab treatment, which appears to be most effective in patients in whom sMAC is elevated. However, as would be predicted from therapy targeted at the TCC, more proximal dysregulation continues unchecked. For this reason, eculizumab treatment appears most appropriate for C3GN patients with elevated sMAC and no or weak C3Nef activity [9]. Additional research including detailed case reports like this one is needed to define the subgroup of DDD/C3GN patients in whom eculizumab therapy can be considered.
As with other glomerular lesions, disease duration is likely to be an important predictor of response to therapy. Eculizumab may have its greatest use in transplant recipients with recurrent disease if it is used either immediately after recognition of recurrence (as in our case) or, as prophylactic therapy initiated immediately post-transplantation (as with aHUS) [25]. As shown in our case, continued therapy with eculizumab slows but does not prevent progression of disease. Prior experience with eculizumab in treating patients with PNH and aHUS suggests that prolonged or even lifelong treatment is required [26, 27].
In conclusion, we present a patient initially diagnosed with DDD who experienced post-transplant recurrent C3GN that partially responded to treatment with eculizumab. The pathophysiological basis of the C3G is more complex than the either PNH or aHUS, both of which are now approved for treatment with eculizumab. Eculizumab treatment should only be offered to C3G patients in the context of a thorough genetic and functional evaluation. In addition, there is a clear need for additional anticomplement therapies that offer the possibility of complement control at the level of the C3 convertase.
References
Habib R, Kleinknecht C, Gubler MC, Maiz HB (1973) Idiopathic membranoproliferative glomerulonephritis. Morphology and natural history. Perspect Nephrol Hypertens 1:491–514
Strife CF, McEnery PT, McAdams AJ, West CD (1977) Membranoproliferative glomerulonephritis with disruption of the glomerular basement membrane. Clin Nephrol 7:65–72
Levy M, Gubler MC, Sich M, Beziau A, Habib R (1978) Immunopathology of membranoproliferative glomerulonephritis with subendothelial deposits (type I MPGN). Clin Immunol Immunopathol 10:477–492
Fakhouri F, Fremeaux-Bacchi V, Noel LH, Cook HT, Pickering MC (2010) C3 glomerulopathy: a new classification. Nat Rev Nephrol 6:494–499
Servais A, Fremeaux-Bacchi V, Lequintrec M, Salomon R, Blouin J, Knebelmann B, Grunfeld JP, Lesavre P, Noel LH, Fakhouri F (2007) Primary glomerulonephritis with isolated C3 deposits: a new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J Med Genet 44:193–199
Sethi S, Fervenza FC, Zhang Y, Nasr SH, Leung N, Vrana J, Cramer C, Nester CM, Smith RJ (2011) Proliferative glomerulonephritis secondary to dysfunction of the alternative pathway of complement. Clin J Am Soc Nephrol 6:1009–1017
Nasr SH, Valeri AM, Appel GB, Sherwinter J, Stokes MB, Said SM, Markowitz GS, D'Agati VD (2009) Dense deposit disease: clinicopathologic study of 32 pediatric and adult patients. Clin J Am Soc Nephrol 4:22–32
Vivarelli M, Pasini A, Emma F (2012) Eculizumab for the treatment of dense deposit disease. N Engl J Med 366:1163–1165
Bomback AS, Smith RJ, Barile GR, Zhang Y, Heher EC, Herlitz L, Stokes MB, Markowitz GS, D'Agati VD, Canetta PA, Radhakrishnan J, Appel GB (2012) Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol 7:748–756
Daina E, Noris M, Remuzzi G (2012) Eculizumab in a patient with dense deposit disease. N Engl J Med 366:1161–1163
McCaughan JA, O'Rourke DM, Courtney AE (2012) Recurrent dense deposit disease after renal transplantation: an emerging role for complementary therapies. Am J Transplant 12:1046–1051
Abrera-Abeleda MA, Nishimura C, Smith JL, Sethi S, McRae JL, Murphy BF, Silvestri G, Skerka C, Jozsi M, Zipfel PF, Hageman GS, Smith RJ (2006) Variations in the complement regulatory genes factor H (CFH) and factor H related 5 (CFHR5) are associated with membranoproliferative glomerulonephritis type II (dense deposit disease). J Med Genet 43:582–589
Zhang Y, Meyer NC, Wang K, Nishimura C, Frees K, Jones M, Katz LM, Sethi S, Smith RJ (2012) Causes of alternative pathway dysregulation in dense deposit disease. Clin J Am Soc Nephrol 7:265–274
Maga TK, Nishimura CJ, Weaver AE, Frees KL, Smith RJ (2010) Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat 31:E1445–E1460
Provan D, Butler T, Evangelista ML, Amadori S, Newland AC, Stasi R (2007) Activity and safety profile of low-dose rituximab for the treatment of autoimmune cytopenias in adults. Haematologica 92:1695–1698
Zaja F, Vianelli N, Volpetti S, Battista ML, Defina M, Palmieri S, Bocchia M, Medeot M, De Luca S, Ferrara F, Isola M, Baccarani M, Fanin R (2010) Low-dose rituximab in adult patients with primary immune thrombocytopenia. Eur J Haematol 85:329–334
Sethi S, Nester CM, Smith RJ (2012) Membranoproliferative glomerulonephritis and C3 glomerulopathy: resolving the confusion. Kidney Int 81:434–441
Martinez-Barricarte R, Heurich M, Valdes-Canedo F, Vazquez-Martul E, Torreira E, Montes T, Tortajada A, Pinto S, Lopez-Trascasa M, Morgan BP, Llorca O, Harris CL, Rodriguez de Cordoba S (2010) Human C3 mutation reveals a mechanism of dense deposit disease pathogenesis and provides insights into complement activation and regulation. J Clin Invest 120:3702–3712
Gale DP, de Jorge EG, Cook HT, Martinez-Barricarte R, Hadjisavvas A, McLean AG, Pusey CD, Pierides A, Kyriacou K, Athanasiou Y, Voskarides K, Deltas C, Palmer A, Fremeaux-Bacchi V, de Cordoba SR, Maxwell PH, Pickering MC (2010) Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet 376:794–801
Habbig S, Mihatsch MJ, Heinen S, Beck B, Emmel M, Skerka C, Kirschfink M, Hoppe B, Zipfel PF, Licht C (2009) C3 deposition glomerulopathy due to a functional factor H defect. Kidney Int 75:1230–1234
Smith RJ, Harris CL, Pickering MC (2011) Dense deposit disease. Mol Immunol 48:1604–1610
Cameron JS, Turner DR, Heaton J, Williams DG, Ogg CS, Chantler C, Haycock GB, Hicks J (1983) Idiopathic mesangiocapillary glomerulonephritis. Comparison of types I and II in children and adults and long-term prognosis. Am J Med 74:175–192
Schena FP, Pertosa G, Stanziale P, Vox E, Pecoraro C, Andreucci VE (1982) Biological significance of the C3 nephritic factor in membranoproliferative glomerulonephritis. Clin Nephrol 18:240–246
Schwertz R, Rother U, Anders D, Gretz N, Scharer K, Kirschfink M (2001) Complement analysis in children with idiopathic membranoproliferative glomerulonephritis: a long-term follow-up. Pediatr Allergy Immunol 12:166–172
Nester C, Stewart Z, Myers D, Jetton J, Nair R, Reed A, Thomas C, Smith R, Brophy P (2011) Pre-emptive eculizumab and plasmapheresis for renal transplant in atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol 6:1488–1494
Parker C (2009) Eculizumab for paroxysmal nocturnal haemoglobinuria. Lancet 373:759–767
Kose O, Zimmerhackl LB, Jungraithmayr T, Mache C, Nurnberger J (2010) New treatment options for atypical hemolytic uremic syndrome with the complement inhibitor eculizumab. Semin Thromb Hemost 36:669–672
Acknowledgments
We are grateful to Helen Mundy and Cheryle Ludwig for assistance with the conduct of this study at the Division of Pediatric Nephrology at UMDNJ. This research was supported, in part, by the National Institutes of Health Grant DK074409 (to R.J.S.).
Disclosures
The investigators were responsible for study design, implementation, data analysis, and manuscript preparation.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Gurkan, S., Fyfe, B., Weiss, L. et al. Eculizumab and recurrent C3 glomerulonephritis. Pediatr Nephrol 28, 1975–1981 (2013). https://doi.org/10.1007/s00467-013-2503-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-013-2503-y