
Abstract
The glomerular basement membrane (GBM) is an especially thick basement membrane that contributes importantly to the kidney’s filtration barrier. The GBM derives from the fusion of separate podocyte and endothelial cell basement membranes during glomerulogenesis and consists primarily of laminin-521 (α5β2γ1), collagen α3α4α5(IV), nidogens-1 and -2, and agrin. Of these nine proteins, mutations in the genes encoding four of them (LAMB2, COL4A3, COL4A4, and COL4A5) cause glomerular disease in humans as well as in mice. Furthermore, mutation of a fifth (Lama5) gene in podocytes in mice causes proteinuria, nephrotic syndrome, and progression to renal failure. These results highlight the importance of the GBM for establishing and maintaining a properly functioning glomerular filtration barrier.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The glomerular filtration barrier between the vasculature and urinary space is tailored to allow the efficient flow of water and small solutes while retarding the passage of plasma proteins, notably albumin and immunoglobulins. Since the first visualization of the glomerular capillary wall’s fine structure by electron microscopy in the 1950s, there has been an ongoing debate among nephrologists, anatomists, pathologists, and molecular cell biologists regarding the origin/mechanism of glomerular permselectivity [1]. In this review I will summarize our current understanding of the cellular and extracellular components of the glomerular capillary wall and focus on the composition of the glomerular basement membrane (GBM) as it relates to permselectivity and kidney disease.
Composition of the glomerular capillary wall
The glomerular capillary wall is a three-layered structure that lies between the vasculature and Bowman’s space and serves as the only barrier between the bloodstream and the urine. Of the three layers, two—the podocyte layer and the endothelial layer—are cellular, whereas the third—the GBM—is a specialized extracellular matrix lying between the two cellular layers. Both podocytes and endothelial cells synthesize and secrete components of the GBM during glomerulogenesis, and both cell types are likely important for maintaining the GBM’s structure and function after glomerular maturation [2–4]. Although the focus of this review is the GBM, each of the three layers will be discussed, as all three are necessary components for establishing and maintaining an intact filtration barrier.
Podocytes
Podocytes, which are also known as glomerular visceral epithelial cells, are anatomically unique epithelial cells that reside within Bowman’s space and are therefore bathed in the primary filtrate. Podocytes enwrap the outer aspect of the glomerular capillaries by extending narrow processes (called “foot processes”) that interdigitate with those of neighboring podocytes. Adjacent foot processes are directly linked to one another by the glomerular slit diaphragms; these derive from molecular modification of the tight junctions that joined the previously columnar, immature podocytes present at earlier stages of glomerulogenesis [5].
Like the tubular epithelial cells of the nephron, podocytes derive from the metanephric mesenchyme, a population of mesoderm-derived cells that become determined to form nephrons during early embryogenesis. At the beginning of meta-nephrogenesis, the epithelial ureteric bud invades the metanephric mesenchyme and induces it to condense and undergo a mesenchyme to epithelium transition to form the renal vesicle. Activation of various signaling pathways results in morphological changes to the renal vesicle and segmentation of the resulting pre-nephron “S-shaped” structure into podocytes, parietal epithelial cells that form Bowman’s capsule, and the tubular segments of the nephron [6].
Glomerular endothelial cells
Like all blood vessels, the glomerular capillaries are lined internally by endothelial cells in direct contact with the bloodstream. However, glomerular endothelial cells are unusual because they bear many fenestrations (from the Latin fenestra, window). Fenestrations are patent holes in the cells that allow the passage of fluid across the endothelial cell layer. It is clear from electron microscopy studies that some of these fenestrations have wagon wheel-like diaphragms similar to those found in peritubular capillaries and in pancreatic islet endothelial cells, while many of the fenestrations do not have diaphragms. The lack of diaphragms likely promotes the flow of plasma across this proximal layer of the glomerular filtration barrier.
Despite the presence of what usually appear under the electron microscope to be wide open fenestrations in the glomerular endothelium, special methods of fixation and staining have revealed the presence of filamentous plugs in the fenestrations. The composition of these plugs is thought to be a form of glycocalyx assembled by the glomerular endothelial cells on their cell surfaces. The plugs have been hypothesized to serve as barriers to the passage of plasma proteins [7–9].
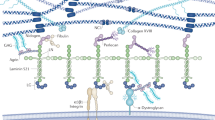
Glomerular basement membrane
The GBM can in some ways be viewed as a typical basement membrane: it has an electron-dense lamina densa when viewed under the electron microscope, and it is formed by associations among the same general classes of macromolecules found in all other types of basement membranes. However, the GBM also has some atypical attributes. It is unusually thick compared to most other basement membranes; this stems from the fact that the GBM forms during glomerulogenesis by the fusion of two distinct basement membranes, the endothelial and the visceral epithelial basement membranes [3]. In addition, the GBM has an unusual composition compared to most other basement membranes, presumably due to its unique functional properties. The GBM contains laminin, type IV collagen, nidogen, and heparan sulfate proteoglycan (HSPG), components found in all basement membranes, but for some of these classes, the specific isoforms present in the GBM are very different than those found in other basement membranes. Even the contiguous Bowman’s capsule and tubular basement membranes, for example, have a very different complement of isoforms compared to the GBM.
Laminin
Laminin describes a family of large, evolutionarily related glycoproteins that assemble with each other in a nonrandom fashion to form at least 15 different αβγ heterotrimeric macromolecules that are secreted by various types of cells into the extracellular space. Laminin trimers are named based on their chain composition; for example, a trimer containing the laminin α2, β1, and γ1 chains is called laminin-211, or LM-211 [10]. Most laminin heterotrimers are cross-shaped structures, with the one “long arm” formed by association of the α, β, and γ chains via coiled-coil interactions and disulfide bonding, and the three “short arms” with NH2-terminal globular domains (called LN domains) that mediate laminin polymerization in the extracellular space. The large, approximately 100kDa laminin globular (LG) domain at the distal end of the long arm (formed exclusively by the COOH-termini of α chains) mediates interactions with receptors on the surface of cells. A number of in vitro and in vivo studies suggest that laminin polymerization in the extracellular space is required for the initiation of basement membrane formation. In addition, laminin polymerization via the short arms is facilitated by LG domain interactions with cellular receptors, perhaps because of the increase in effective laminin concentration that results near the cell surface. Intracellular signaling events induced by laminin binding are also involved in promoting laminin polymerization and basement membrane formation [11].
The major laminin trimer found in the mature GBM is LM-521 (α5β2γ1). LM-521 is secreted by both podocytes and endothelial cells [2]. At early stages of glomerulogenesis, when the glomerular endothelial cells migrate towards the presumptive podocytes to begin to form the glomerular capillaries and the GBM is first recognizable as such, LM-111 and LM-511 are the major laminin components [12]. However, as the capillaries begin to mature, LM-521 begins to be deposited by the podocytes and endothelial cells, and LM-111 and -511 are eventually eliminated [12, 13]. The molecular mechanisms responsible for these developmental transitions have yet to be discovered, but as mentioned below they are crucial to ensure proper permselectivity.
Type IV Collagen
Like all collagens, type IV collagens are secreted as heterotrimers (also called protomers) composed of three α chains whose sequences include collagenous domains consisting of many Gly-X-Y amino acid triplet repeats. The presence of glycine at every third residue allows the chains to assemble into a triple helix, as glycine is the only amino acid that can fit at the center of the helix. However, unlike the rigid forms of collagen that are associated with bone, for example, collagen IV α chains contain multiple interruptions of the Gly-X-Y repeats scattered throughout the collagenous domain. These interruptions are thought to provide flexibility to the collagen IV trimers and therefore to the collagen IV network, which is formed by covalent and non-covalent interactions among trimers [14].
Aside from the central collagenous domain containing the interruptions, all six collagen IV α chains also have a small noncollagenous NH2-terminal domain called 7S and a larger COOH-terminal noncollagenous domain called NC1. Both of these domains are involved in linking trimers to each other to promote collagen IV network formation. In addition, the NC1 domain is crucial for directing the composition of collagen IV heterotrimers; information in the form of sequence and structure embedded in the NC1 domain ensures that only three types of trimers form: (α1)2α2, α3α4α5, and (α5)2α6 [15].
The collagen IV network of the mature GBM is composed primarily of α3α4α5 heterotrimers, although there is also some (α1)2α2 that is detectable by immunohistochemical methods, especially at the GBM’s endothelial aspect. Consistent with this, podocytes—but not endothelial cells—synthesize the α3α4α5 network [4], so it is likely the α1 and α2 chains that are present in the GBM derive from the glomerular endothelial cells. As with laminins, there are also developmental transitions in collagen IV gene expression during glomerulogenesis. At early stages, only α1 and α2 chains are detected, but as the glomerular capillaries begin to mature, there is a gradual increase in deposition of the α3, α4, and α5 chains [13]. This developmental transition in collagen IV network expression has been shown to occur during human, dog, and mouse kidney development [13, 16, 17].
Nidogens
Nidogens-1 and -2 are dumbbell-shaped basement membrane proteins that bind tightly to the laminin γ1 chain short arm and also bind to type IV collagen [18]. Thus, nidogen was initially thought to be crucial for basement membrane formation as a linker between the separate laminin and type IV collagen networks. However, the generation and analysis of single and double nidogen-1 and -2 knockout mice show that basement membranes can form in the total absence of nidogen, although some basement membranes that are subject to increased stress during development exhibit impaired integrity in the absence of both nidogens [19]. Both nidogen-1 and -2 are present in the GBM, but little is known about their developmental expression patterns.
Heparan sulfate proteoglycans
Heparan sulfate proteoglycans consist of a protein core with covalently linked glycosaminoglycan side chains. These side chains are frequently modified by sulfation, which imparts a negative charge to the proteoglycan. Although perlecan is likely the most common HSPG found in basement membranes, the mature GBM contains primarily agrin [20]. Agrin is best known for its role in the neuromuscular system, where a particular splice form secreted by motor neurons is crucial for the development and organization of neuromuscular junctions [21]. Although agrin is the major GBM HSPG at maturity, during glomerulogenesis there appears to be co-distribution of perlecan and agrin, following which perlecan disappears and becomes confined to the mesangial matrix [22].
The GBM’s contribution to permselectivity
Since the identification of mutations in nephrin, a component of the glomerular slit diaphragm, in congenital nephrotic syndrome of the Finnish type [23], podocytes have become a major focus of research aimed at elucidating the nature of the glomerular filtration barrier. There are now known to be numerous genes expressed in podocytes whose mutation in humans and/or mice results in impaired glomerular permselectivity and nephrotic range proteinuria [24]. This certainly validates the importance of podocytes for establishing and maintaining the glomerular filtration barrier. However, the GBM should not be ignored or forgotten [25]. (The same holds true for the endothelial glycocalyx [9], but that is beyond the scope of this review.) Indeed, of the nine known matrix proteins present in the mature GBM, mutations that affect four of them cause glomerular disease in humans, and mutation of a fifth causes nephrotic syndrome in mice [26]. These will now be discussed.
The first GBM protein shown to be affected in a human disease is collagen α5(IV) [27]. Mutations in COL4A5 cause the X-linked (and most common) form of Alport syndrome, a hereditary glomerulonephritis leading to end-stage kidney disease that is accompanied by hearing loss and lens defects [28]. As discussed above, the α5 chain of type IV collagen is a component of the α3α4α5(IV) trimer that is present in the GBM. In the absence of α5(IV), these trimers and the network they would normally form within the GBM are absent. Likewise, homozygous mutations that affect COL4A3 or COL4A4 also prevent production of the α3α4α5(IV) trimer and cause the autosomal recessive form of Alport syndrome. In the absence of the usual GBM collagen IV network, there is substitution or compensation by the (α1)2α2(IV) network throughout the width of the GBM [29]. Although this network functions adequately for several years in Alport patients, eventually there is splitting and thickening of the GBM associated first with hematuria and later with proteinuria. Given that, it seems that the identity of the collagen IV network may not be an important determinant of glomerular permselectivity. It is worth mentioning here that dominant mutations in human COL4A1 also cause several forms of kidney disease with diverse extrarenal manifestations [30, 31].
In contrast to the delayed onset proteinuria in Alport syndrome, patients with Pierson syndrome exhibit congenital nephrotic syndrome, as well as extrarenal manifestations, such as small pupils and neurological abnormalities [32]. Pierson syndrome is caused by truncating or severe missense mutations in LAMB2 [33], which encodes the laminin β2 chain. Lamb2-/- mice, which lack detectable laminin β2, have nephrotic syndrome [34], severe neuromuscular abnormalities [35], and retinal defects [36], all of which are consistent with the features of Pierson syndrome. Patients with some apparently less severe missense LAMB2 mutations have nephrotic syndrome and significantly milder extrarenal defects [37, 38]. These data suggest that laminin β2 and the laminin-521 trimer of which it is normally a part in the GBM contribute directly to the filtration barrier. Studies in Lamb2-/- mice at different ages showed that proteinuria precedes foot process effacement and the loss of slit diaphragms, indicating that the GBM was indeed defective and that it normally serves as an important component of the filtration barrier, whereas loss of foot processes seems to be a secondary response to proteinuria [39].
Regarding the other components of the GBM, lack of either nidogen-1 or nidogen-2 results in viable mice [19] and no significant defects in glomerular filtration. Similarly, lack of agrin in the GBM has no discernible effect on filtration, despite the fact that its loss causes a dramatic reduction in the GBM’s concentration of anionic sites [22]. Finally, although no mutations affecting human laminin α5 or laminin γ1 have been discovered, likely due to early prenatal lethality, podocyte-specific mutation of Lama5 in mice results in proteinuria that in many cases progresses to nephrotic syndrome and renal failure [26].
References
Farquhar MG (1975) Editorial: The primary glomerular filtration barrier–basement membrane or epithelial slits? Kidney Int 8:197–211
St. John PL, Abrahamson DR (2001) Glomerular endothelial cells and podocytes jointly synthesize laminin-1 and -11 chains. Kidney Int 60:1037–1046
Abrahamson DR (1985) Origin of the glomerular basement membrane visualized after in vivo labeling of laminin in newborn rat kidneys. J Cell Biol 100:1988–2000
Abrahamson DR, Hudson BG, Stroganova L, Borza DB, St John PL (2009) Cellular origins of type IV collagen networks in developing glomeruli. J Am Soc Nephrol 20:1471–1479
Schnabel E, Anderson JM, Farquhar MG (1990) The tight junction protein ZO-1 is concentrated along slit diaphragms of the glomerular epithelium. J Cell Biol 111:1255–1263
Dressler GR (2006) The Cellular Basis of Kidney Development. Annu Rev Cell Dev Biol 22:509–529
Deen WM, Lazzara MJ, Myers BD (2001) Structural determinants of glomerular permeability. Am J Physiol Ren Physiol 281:F579–F596
Haraldsson B, Nystrom J, Deen WM (2008) Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev 88:451–487
Haraldsson B, Jeansson M (2009) Glomerular filtration barrier. Curr Opin Nephrol Hypertens 18:331–335
Aumailley M, Bruckner-Tuderman L, Carter WG, Deutzmann R, Edgar D, Ekblom P, Engel J, Engvall E, Hohenester E, Jones JC, Kleinman HK, Marinkovich MP, Martin GR, Mayer U, Meneguzzi G, Miner JH, Miyazaki K, Patarroyo M, Paulsson M, Quaranta V, Sanes JR, Sasaki T, Sekiguchi K, Sorokin LM, Talts JF, Tryggvason K, Uitto J, Virtanen I, von der Mark K, Wewer UM, Yamada Y, Yurchenco PD (2005) A simplified laminin nomenclature. Matrix Biol 24:326–332
Colognato H, Winkelmann DA, Yurchenco PD (1999) Laminin polymerization induces a receptor-cytoskeleton network. J Cell Biol 145:619–631
Miner JH, Patton BL, Lentz SI, Gilbert DJ, Snider WD, Jenkins NA, Copeland NG, Sanes JR (1997) The laminin alpha chains: expression, developmental transitions, and chromosomal locations of alpha1-5, identification of heterotrimeric laminins 8–11, and cloning of a novel alpha3 isoform. J Cell Biol 137:685–701
Miner JH, Sanes JR (1994) Collagen IV α3, α4, and α5 chains in rodent basal laminae: Sequence, distribution, association with laminins, and developmental switches. J Cell Biol 127:879–891
Hudson BG (2004) The molecular basis of Goodpasture and Alport syndromes: beacons for the discovery of the collagen IV family. J Am Soc Nephrol 15:2514–2527
Khoshnoodi J, Cartailler JP, Alvares K, Veis A, Hudson BG (2006) Molecular recognition in the assembly of collagens: terminal noncollagenous domains are key recognition modules in the formation of triple helical protomers. J Biol Chem 281:38117–38121
Harvey SJ, Zheng K, Sado Y, Naito I, Ninomiya Y, Jacobs RM, Hudson BG, Thorner PS (1998) Role of distinct type IV collagen networks in glomerular development and function. Kidney Int 54:1857–1866
Kalluri R, Shield CF III, Todd P, Hudson BG, Neilson EG (1997) Isoform switching of type IV collagen is developmentally arrested in X-linked Alport syndrome leading to increased susceptibility of renal basement membranes to endoproteolysis. J Clin Invest 99:2470–2478
Salmivirta K, Talts JF, Olsson M, Sasaki T, Timpl R, Ekblom P (2002) Binding of mouse nidogen-2 to basement membrane components and cells and its expression in embryonic and adult tissues suggest complementary functions of the two nidogens. Exp Cell Res 279:188–201
Bader BL, Smyth N, Nedbal S, Miosge N, Baranowsky A, Mokkapati S, Murshed M, Nischt R (2005) Compound genetic ablation of nidogen 1 and 2 causes basement membrane defects and perinatal lethality in mice. Mol Cell Biol 25:6846–6856
Groffen AJ, Ruegg MA, Dijkman H, van de Velden TJ, Buskens CA, van den Born J, Assmann KJ, Monnens LA, Veerkamp JH, van den Heuvel LP (1998) Agrin is a major heparan sulfate proteoglycan in the human glomerular basement membrane. J Histochem Cytochem 46:19–27
Kummer TT, Misgeld T, Sanes JR (2006) Assembly of the postsynaptic membrane at the neuromuscular junction: paradigm lost. Curr Opin Neurobiol 16:74–82
Harvey SJ, Jarad G, Cunningham J, Rops AL, van der Vlag J, Berden JH, Moeller MJ, Holzman LB, Burgess RW, Miner JH (2007) Disruption of glomerular basement membrane charge through podocyte-specific mutation of agrin does not alter glomerular permselectivity. Am J Pathol 171:139–152
Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan CE, Peltonen L, Holmberg C, Olsen A, Tryggvason K (1998) Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol Cell 1:575–582
Machuca E, Benoit G, Antignac C (2009) Genetics of nephrotic syndrome: connecting molecular genetics to podocyte physiology. Hum Mol Genet 18:R185–R194
Farquhar MG (2006) The glomerular basement membrane: not gone, just forgotten. J Clin Invest 116:2090–2093
Goldberg S, Adair-Kirk TL, Senior RM, Miner JH (2010) Maintenance of glomerular filtration barrier integrity requires laminin alpha5. J Am Soc Nephrol 21:579–586
Barker DF, Hostikka SL, Zhou J, Chow LT, Oliphant AR, Gerken SC, Gregory MC, Skolnick MH, Atkin CL, Tryggvason K (1990) Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 248:1224–1227
Kashtan CE (2000) Alport syndromes: phenotypic heterogeneity of progressive hereditary nephritis. Pediatr Nephrol 14:502–512
Kashtan CE, Kim Y (1992) Distribution of the α1 and α2 chains of collagen IV and of collagens V and VI in Alport syndrome. Kidney Int 42:115–126
Gould DB, Phalan FC, van Mil SE, Sundberg JP, Vahedi K, Massin P, Bousser MG, Heutink P, Miner JH, Tournier-Lasserve E, John SW (2006) Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J Med 354:1489–1496
Plaisier E, Gribouval O, Alamowitch S, Mougenot B, Prost C, Verpont MC, Marro B, Desmettre T, Cohen SY, Roullet E, Dracon M, Fardeau M, Van Agtmael T, Kerjaschki D, Antignac C, Ronco P (2007) COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N Engl J Med 357:2687–2695
Zenker M, Tralau T, Lennert T, Pitz S, Mark K, Madlon H, Dotsch J, Reis A, Muntefering H, Neumann LM (2004) Congenital nephrosis, mesangial sclerosis, and distinct eye abnormalities with microcoria: an autosomal recessive syndrome. Am J Med Genet A 130:138–145
Zenker M, Aigner T, Wendler O, Tralau T, Muntefering H, Fenski R, Pitz S, Schumacher V, Royer-Pokora B, Wuhl E, Cochat P, Bouvier R, Kraus C, Mark K, Madlon H, Dotsch J, Rascher W, Maruniak-Chudek I, Lennert T, Neumann LM, Reis A (2004) Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet 13:2625–2632
Noakes PG, Miner JH, Gautam M, Cunningham JM, Sanes JR, Merlie JP (1995) The renal glomerulus of mice lacking s-laminin/laminin β2: nephrosis despite molecular compensation by laminin β1. Nat Genet 10:400–406
Noakes PG, Gautam M, Mudd J, Sanes JR, Merlie JP (1995) Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/laminin β2. Nature 374:258–262
Libby RT, Lavallee CR, Balkema GW, Brunken WJ, Hunter DD (1999) Disruption of laminin beta2 chain production causes alterations in morphology and function in the CNS. J Neurosci 19:9399–9411
Hasselbacher K, Wiggins RC, Matejas V, Hinkes BG, Mucha B, Hoskins BE, Ozaltin F, Nurnberg G, Becker C, Hangan D, Pohl M, Kuwertz-Broking E, Griebel M, Schumacher V, Royer-Pokora B, Bakkaloglu A, Nurnberg P, Zenker M, Hildebrandt F (2006) Recessive missense mutations in LAMB2 expand the clinical spectrum of LAMB2-associated disorders. Kidney Int 70:1008–1012
Matejas V, Hinkes B, Alkandari F, Al-Gazali L, Annexstad E, Aytac MB, Barrow M, Blahova K, Bockenhauer D, Cheong HI, Maruniak-Chudek I, Cochat P, Dotsch J, Gajjar P, Hennekam RC, Janssen F, Kagan M, Kariminejad A, Kemper MJ, Koenig J, Kogan J, Kroes HY, Kuwertz-Broking E, Lewanda AF, Medeira A, Muscheites J, Niaudet P, Pierson M, Saggar A, Seaver L, Suri M, Tsygin A, Wuhl E, Zurowska A, Uebe S, Hildebrandt F, Antignac C, Zenker M (2010) Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum. Hum Mutat 31:992–1002
Jarad G, Cunningham J, Shaw AS, Miner JH (2006) Proteinuria precedes podocyte abnormalities in Lamb2-/- mice, implicating the glomerular basement membrane as an albumin barrier. J Clin Invest 116:2272–2279
Acknowledgments
My relevant research is supported by NIH grants R01DK078314, R01GM060432, and R01DK081156.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Miner, J.H. Glomerular basement membrane composition and the filtration barrier. Pediatr Nephrol 26, 1413–1417 (2011). https://doi.org/10.1007/s00467-011-1785-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-011-1785-1