
Abstract
Chronic kidney diseases (CKD), independent of their primary cause, lead to progressive, irreversible loss of functional renal parenchyma. Renal pathology in CKD is characterized by tubulointerstitial fibrosis with excessive matrix deposition produced by myofibroblasts. Because blocking the formation of these scar-forming cells represents a logical therapeutic target for patients with progressive fibrotic kidney disease, the origin of renal myofibroblasts is a subject of intense investigation. Although the traditional view holds that resident fibroblasts are the myofibroblast precursor, for the last 10 years, injured epithelial cells have been thought to directly contribute to the myofibroblast pool by the process of epithelial-to-mesenchymal transition (EMT). The recent application of genetic fate mapping techniques in mouse fibrosis models has provided new insights into the cell hierarchies in fibrotic kidney disease and results cast doubt on the concept that EMT is a source of myofibroblast recruitment in vivo, but rather point to the resident pericyte/perivascular fibroblast as the myofibroblast progenitor pool. This review will highlight recent findings arguing against EMT as a direct contributor to the kidney myofibroblast population and review the use of genetic fate mapping to elucidate the cellular mechanisms of kidney homeostasis and disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Renal fibrosis is considered a maladaptive repair process and its hallmarks include chronic inflammation, a persistent irritant that sustains the generation of growth factors, fibrogenic cytokines and proteolytic enzymes, and recruitment and proliferation of myofibroblasts that synthesize and deposit extracellular matrix leading to progressive remodeling and destruction of normal kidney tissue architecture [1]. The repair process can be subdivided into two phases: the first “regenerative” phase is characterized by an attempt to replace injured cells by cells of the same type which would lead to full recovery of kidney parenchyma (restitutio ad integrum) leaving no lasting evidence of damage. Depending on the severity, duration, and type of injury, this initially beneficial reaction can however be gradually followed by a phase called fibroplasia or fibrosis, in which functional tissue is remodeled and subsequently replaced by connective tissue, resulting in irreversible scar formation [2]. In kidney, this is reflected by a progressive expansion of the tubulointerstitium and demise of adjacent cellular structures leading to nephron loss and ultimately end-stage kidney disease. Not surprisingly, the extent of tubulointerstitial damage in any given biopsy is inversely correlated with renal function and has good predictive value for kidney survival—be it native or allograft [3, 4].
Scientists across a wide range of disciplines agree that myofibroblasts directly lay down pathological matrix components that constitute fibrotic scarring and are therefore an attractive target cell type for anti-fibrotic therapeutic intervention. While virtually absent in healthy kidney, myofibroblasts are abundantly found in active renal fibrosis. Clarifying the origin of this cell population, uncovering mechanisms of recruitment, and understanding their biological properties are essential for the identification of potential targets and development of new therapies to treat chronic kidney diseases.
Fibroblasts and myofibroblasts: definition and background
Activated fibroblasts or myofibroblasts are generally accepted as the key effector cell in the pathogenesis of fibrosis in kidney and other solid organs. Identifying fibroblasts in the kidney can be challenging, and usually requires a combination of criteria such as cell localization, morphology, and marker expression. Fibroblasts are spindle-shaped cells of mesenchymal origin that are surrounded by (and give rise to) connective tissue. Several markers have been proposed for labeling renal fibroblasts including nerve growth factor, platelet-derived growth factor receptors α and β, CD73 (ecto-5′-nucleotidase), CD90 (a cell adhesion glycoprotein and member of the immunoglobulin superfamily), and a calcium-binding protein S100A4 also known as fibroblast-specific protein-1 (FSP-1), although FSP-1 is not in fact specific to fibroblasts [5].
The activation of a “quiescent” fibroblast and transition to a myofibroblast in fibrogenesis is classically marked in rodents by de novo expression of α-smooth muscle actin (αSMA) [6, 7] consistent with a role for these cells in producing contractile forces characteristic of connective-tissue remodeling [8]. Most, but not all, myofibroblasts across organs express αSMA [9], and αSMA expression is regulated in concert with type I collagen by cytokines including transforming growth factor-β1 [10], emphasizing a role for myofibroblasts in both extracellular matrix deposition and contraction. Expression of other matrix proteins such as the ED-A splice variant of fibronectin is also associated with myofibroblast differentiation [11]. These activated cells are typically highly proliferative, and recent evidence suggests that myofibroblast activation may lead to permanently upregulated proliferative activity and matrix synthesis capacity through epigenetic changes [12]. The molecular anatomy of myofibroblasts is not fully understood, and one open question is whether myofibroblasts may be a more heterogeneous population than originally thought.
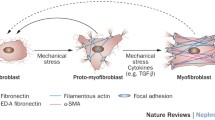
The origin of myofibroblasts has long been a subject of investigation and debate (Fig. 1). While investigating mechanisms of inflammation, Julius Friedrich Cohnheim, a pioneer of experimental pathology, published a comprehensive article in 1867, characterizing so-called “contractile cellular elements” or fibroblasts as descendents of migrating white blood cells [13]. It was not until 1970 that this paradigm would shift when Russell Ross and colleagues provided strong evidence obtained from a set of elegant experiments using parabiotic rats that fibroblasts were unlikely to arise from hematogenous precursors but were rather of local origin [14]. Until the late 1980s, resident interstitial fibroblasts were considered the only source of extracellular matrix in mature tissue. Since that time, the notion that epithelial cells (under conditions of chronic injury) undergo a mesenchymal transition (epithelial to mesenchymal transition or EMT) and traverse the basement membrane to become interstitial myofibroblasts has gained wide acceptance [15, 16]. Recently, however, genetic fate mapping has called the EMT hypothesis into question. This new data will be reviewed (Fig. 1).

Current concepts of myofibroblast recruitment in CKD. The traditional concept holds that myofibroblasts primarily derive from local stromal cells in the kidney such as resident fibroblasts and pericytes/perivascular fibroblasts. Epithelial-mesenchymal transition (EMT), a process in which epithelial cells are believed to undergo complete phenotypical transformation, acquire mesenchymal properties and traverse the tubular basement membrane (TBM), has been proposed as an important alternative route of recruitment. Recent studies using state-of-the-art fate mapping techniques, however, have cast serious doubt on the significance of EMT as generator of myofibroblasts in renal fibrosis. Other proposed sources of recruitment include endothelial cells (via endothelial-mesenchymal transition, EndMT) and bone-marrow-derived cells. Published data indicates that bone-marrow-derived cells make very little, if any, contribution to collagen-secreting cells in kidney fibrosis
Evidence supporting an epithelial origin for kidney myofibroblasts
The process of EMT was originally described as an early event in embryogenesis conferring migratory capacity to primitive epithelia to form the mesoderm and enabling primitive neuroepithelia to move as neural crest cells [17, 18]. In the field of cancer biology, EMT is an important molecular program for tumor invasion and metastasis [19]. However, it should be noted that cancer EMT does not result in the formation of metastatic lesions comprised of fibroblasts, but rather epithelial cells with mesenchymal characteristics that are clearly distinct from fibroblasts. For the last decade, a growing volume of data suggests that the process of EMT may also represent a major direct route of myofibroblast recruitment during fibrogenesis [20]. This model, also referred to as type 2 EMT, holds that adult epithelial cells, under conditions of chronic injury that change the composition of local cytokines, undergo dedifferentiation, traverse the tubular basement membrane into the interstitium, and finally “transdifferentiate” into a myofibroblast phenotype capable of synthesizing and depositing extracellular matrix. Evidence to support this hypothesis rests on several lines of inquiry. First, many studies report that primary tubular epithelial cells when cultured in the presence of fibrogenic TGF-β1 in vitro can undergo phenotypic conversion. Characteristic features of this transition include loss of epithelial markers such as E-cadherin, zonula occludens-1 (ZO-1) and cytokeratin, activation of mesenchymal or myofibroblast marker expression such as vimentin, αSMA, FSP-1 and interstitial matrix components as well as adoption of a spindle-like morphology [21]. Second, immunohistological analysis of injured kidney has demonstrated co-localization of epithelial and mesenchymal markers suggesting an intermediate cell-type captured in transition from epithelial cell to fibroblast. Finally, and most importantly, in a landmark article published in 2002, the first direct evidence for EMT in vivo was presented utilizing Cre/Lox technology and cell lineage analysis to track the cell fate of renal epithelial cells during fibrotic disease [22]. In this study, the transgene expressing Cre recombinase under regulation of the γ-Glutamyl Transferase (γGT) promoter was expressed in the Rosa26 LacZ reporter mouse (R26R). γGT is expressed in proximal tubule. Hence LacZ is permanently activated in proximal tubule and can be used to monitor the migration of tubular epithelium and its progeny in kidney disease. Applying the unilateral ureteral obstruction (UUO) model of progressive renal fibrosis, LacZ-positive cells could be detected in the tubulointerstitium by β-gal immunostaining, and the authors reported that ∼36% of all FSP1+ interstitial fibroblasts derived from proximal tubular epithelium via EMT. However, no confirmatory study in the kidney using lineage analysis has been published.
Techniques for identification of myofibroblast progenitors
A full discussion of the origin of renal myofibroblasts and the role of EMT requires review of the methods used to identify myofibroblast origin and to define EMT. Three primary methods have been used for identifying kidney myofibroblast progenitor cells. First, surrogate markers of EMT have been investigated in vivo during renal fibrosis, correlating expression of presumed EMT markers in epithelial and/or myofibroblast cell types. Demonstrating motility (for example of epithelial cells in transit to the interstitium) in vivo is difficult. As a consequence, markers of EMT have been adopted as surrogates for the process by which an epithelial cell turns into a fibroblast. The typical markers used include loss of epithelial markers such as E-cadherin and gain of mesenchymal markers such as vimentin, αSMA, or S100A4/FSP1. Unfortunately, the loss of E-cadherin may simply reflect epithelial injury with loss of cell polarity. Similarly, neither gain of vimentin nor S100A4/FSP-1 expression specifies an EMT process. Indeed, vimentin expression has long been known to be involved in the dedifferentiation and mitogenic response of viable epithelial cells after ischemic injury to the kidney, and thus de facto participates in regenerative processes of the tubular epithelium [23, 24]. S100A4/FSP-1 has been shown, in carefully performed studies, not to be detectable in myofibroblasts or cells that generate collagen matrix in rodent kidney, but rather in leukocytes [25–27], suggesting that the label “fibroblast-specific protein” is inaccurate.
Second, a variety of cell-culture models have been used to examine the process by which epithelial cells can undergo EMT, losing their epithelial characteristics and gaining expression of mesenchymal proteins. These in vitro studies, which are most numerous in the literature, rest on the assumption that because EMT can be observed in a Petri dish that it must also occur in vivo. However, as we shall review, the ability of epithelial cells to express mesenchymal proteins in vitro is well established, but has little bearing on whether epithelial cells adopt an alternative cell identity (that of the myofibroblast) in vivo.
Third, the most powerful technique applied to this question is fate mapping, a method of defining a cell’s history back to an earlier stage and traditionally used in developmental biology. Combining modern genetic manipulation techniques with traditional fate-mapping approaches has created genetic fate-mapping, a powerful technique that combines cell labeling at molecular precision with the unambiguous detection of its progeny. More recently, this technique has been applied to adult organs in order to clarify lineage relationships in complex tissues in both development and disease and it represents the gold standard technique for identifying a cell’s progenitor [28, 29]. Genetic fate mapping utilizes a site-specific recombinase—an enzyme that recognizes specific nucleotide sequences to mediate DNA excision—to activate expression of a reporter molecule such as green fluorescent protein (GFP). In the absence of recombinase activity, a DNA cassette upstream of the reporter prevents its expression. This “STOP” cassette is flanked by nucleotide sequences specifically recognized by the recombinase and expression of the recombinase leads to cassette excision and re-ligation of DNA strands with activation of constitutive reporter expression. The label is heritable because the reporter is integrated into the genome and is regulated by promoter/enhancers that are active in all cells. Therefore, both the ancestor and all descendent cells continue to express it. Bacterial Cre recombinase (cyclization recombination) in combination with LoxP nucleotide recognition sites (locus of x-over P1, consisting of 34 base pairs 5′-ATAACTTCGTATA-GCATACAT-TATACGAAGTTAT-3′) is most commonly used in mammalian fate mapping, and advanced variants exist including one in which a modified version of the estrogen receptor ligand-binding domain is fused (CreER). In the absence of a ligand, the CreER recombinase is unable to access the eukaryotic nucleus, but administration of tamoxifen or its active derivative 4-hydroxytamoxifen (4-OHT) allows nuclear translocation where recombination can occur, permitting temporal control over the genetic marking process [30].
Recent studies argue against any contribution of epithelial cells to the myofibroblast pool
While a multitude of studies have provided indirect support for the existence of EMT through induced expression of mesenchymal proteins by epithelial cells, only Iwano et al. utilized genetic fate mapping to investigate EMT in vivo, with no follow-up fate-mapping studies [22]. Taking advantage of the development of new Cre-driver mice and improved lineage reporters, we recently re-examined the question of whether kidney myofibroblasts derive from epithelial cells. We genetically labeled all renal epithelial cells in kidney using two separate and well-characterized developmentally active Cre-driver lines: the Six2-cre driver, which labels metanephric mesenchyme-derived epithelia [31], and the HoxB7-cre driver, which labels ureteric bud-derived collecting duct epithelia [32]. Two different reporter lines were used, the Rosa26 Reporter (R26R) (with bacterial LacZ gene as fate marker) and the Z/Red (with DsRed as a reporter) mouse lines. Bigenic mice containing both the Cre and reporter alleles demonstrated strong expression in either LacZ or DsRed exclusively in epithelial compartments, as expected. To examine the fate of these lineage-marked epithelial cells, we used two models of fibrotic disease—unilateral ureteral obstruction (UUO) and unilateral ischemia-reperfusion injury, to induce kidney fibrosis. Despite the robust appearance of aSMA-positive interstitial myofibroblasts, we were unable to detect genetically labeled cells in the interstitium; that is cells derived from kidney epithelium. By contrast, when we cultured primary renal epithelial cells in vitro from these mice and incubated them with TGF-β, the DsRed-positive epithelial cells readily expressed aSMA and lost E-cadherin expression, a reported characteristic of EMT. These experiments support the notion that an EMT-like process occurs in vitro, whereby epithelial cells lose markers of terminal differentiation and gain expression of mesenchymal markers, but they provide strong evidence against the idea that epithelial cells turn into myofibroblasts, traverse the basement membrane, and take up residence in the interstitium—a cornerstone of the EMT hypothesis.
Four other studies utilizing genetic fate mapping in fibrotic renal models have been reported since our publication and they also fail to find support for the EMT hypothesis. Li et al. crossed the ksp-cadherin-Cre driver with the R26-EYFP reporter to generate YFP-tagged renal epithelia and subjected the mice to UUO [27]. No interstitial YFP-labeled myofibroblasts were observed in the interstitium. The ksp-cadherin-Cre driver labeled collecting duct, distal tubule and loop epithelium well but fate-marked only 21% of the proximal tubule epithelium. The authors therefore conducted additional experiments labeling proximal tubules with Texas-Red-conjugated dextran. While proximal tubule epithelia retained dextran following UUO, no dextran-labeled interstitial cells were detected—confirming that EMT did not generate interstitial myofibroblasts in the UUO model. Endo and colleagues developed a novel proximal tubule-specific CreERt2 knockin to the proximal tubule-specific gene N-myc downstream-regulated gene-1 (NDRG1). After crossing the NDRG1-CreERt2 driver to an eGFP reporter mouse, they genetically labeled over 80% of proximal tubule epithelia with sequential tamoxifen pulses—a much higher fraction than was labeled in the study by Li et al. which would have increased the sensitivity to detect EMT if it were occurring. Similar to our results and those of Li et al., however, Endo and colleagues found no eGFP-positive interstitial myofibroblasts after 14 days of ureteral obstruction [33].
Koesters and colleagues utilized the inducible expression of transforming growth factor (TGF)-β in renal epithelia to examine epithelial fate in renal fibrosis. This elegant model consists of a Pax8 promoter driving tubular epithelial expression of the reverse tetracycline transactivator combined with tetracycline-responsive elements upstream of both TGF-β and Cre, as well as a R26R allele. In this quadruple transgenic, the addition of doxycycline simultaneously activates epithelial TGF-β overexpression while fate marking the same TGF-β-producing cell with the LacZ reporter. The authors observed focal proliferation of peritubular fibroblasts with collagen deposition around tubules where TGF-β overexpression occurred, as defined by both TGF-β staining and LacZ expression. They did not observe these same epithelial cells transitioning into myofibroblasts, and when they examined the fate of the lineage-marked tubular cells that overexpressed TGF-β, they did not identify cells transgressing the basement membrane nor did they detect them in the tubulointerstitium. Instead, they observed decomposition of tubular epithelia leaving a denuded, cell-free tubule with intact basement membrane and surrounded by fibrosis [34]. Since TGF-β has been considered a master regulator of EMT [35], it is especially noteworthy that this study failed to detect any contribution of EMT to interstitial fibrosis in this strictly TGF-β-driven model. Finally, Bielesz and colleagues labeled renal epithelial cells with a PEPCK-Cre driver crossed to the R26R, and using a folic acid nephropathy fibrosis model, these investigators, “failed to find epithelial cells in the interstitium [36].”
Thus, since our own report, four additional investigations have been published using the gold standard assay for tracking cell fate in vivo—genetic fate mapping—and all studies have concluded that EMT does not result in the generation of interstitial myofibroblasts in fibrotic renal disease. This conclusion is strengthened by the variety of independent Cre drivers (six) and reporters (four) utilized, the inclusion of positive and negative controls for fate-marker detection and the use of four different fibrotic injury models (UUO, ischemia reperfusion injury, folate nephropathy and TGF-β overexpression) by five independent laboratories that all arrived at the same conclusion. The reason for the discrepant results between Iwano et al. on the one side, and our study and the others’ summarized here on the other, are unknown. Perhaps importantly, the original fate-mapping result from Iwano et al. that supports an epithelial origin for some myofibroblasts [22] was based on immunofluorescent antibody detection of the β-gal product of the LacZ genetic fate marker driven by the R26 locus. We and others have been unable to detect with confidence LacZ expressed by the Rosa26 promoter in adult kidney cortex epithelia by immunofluorescence [32, 37]. Positive and negative controls for fluorescence marker detection in kidney are required to set the signal threshold above which a cell will be counted as positive—i.e., to distinguish cells expressing the fate marker from autofluorescence—but there is no mention of these controls in the 2002 report. A similar controversy has existed in cardiac regeneration, where an early report showing bone marrow transdifferentiation into cardiac myocytes [38] was later shown to most likely reflect the high intrinsic autofluorescence in myocardium [39, 40]. Ultimately reconciling these conflicting reports may be very challenging: these findings have stirred debate [41] and will no doubt continue to do so [42].
It should be noted that a variety of other studies using more traditional techniques have also failed to find evidence implicating EMT in the generation of interstitial myofibroblasts. Using a model of anti-glomerular basement membrane antibody-induced crescentic nephritis, Wiggins and colleagues demonstrated an increase of collagen I expression in interstitial cells with close proximity to vessels calling them “vascular adventitial” or “periadventitial” cells [43]. Corroborating observations were reported by Faulkner et al. in an accelerated model of angiotensin II-induced renal fibrosis following habu venom injury [44]. The authors noticed an early expansion of αSMA-positive myofibroblasts in perivascular regions and determined that the vast majority of tubulointerstitial αSMA-expressing cells had to be of local origin, but found no evidence for proximal epithelial cells translocating into the tubulointerstitium by labeling with Texas Red dextran. Lin and coworkers used a reporter mouse model expressing GFP under the control of the collagen I, alpha 1 (coll1α1) promoter to study the origins of coll1α1-producing cells in the kidney. Using time course microscopy and kinetic modeling, they identified the initial population of pericytes and perivascular fibroblasts as the primary source of myofibroblasts in the fibrotic kidney [26]. Finally, Picard et al. performed careful co-labeling studies of resident fibroblasts at early time-points after ureteral obstruction, and observed very early proliferation and acquisition of αSMA expression in resident fibroblasts without any evidence for EMT [45].
Doubt regarding the EMT hypothesis has also been raised by a number of renal pathologists who fail to catch epithelial cells in the actual act of traversing the basement membrane—an event that one would expect to routinely witness in active fibrosis [46]. While those who favor EMT argue that these cells may be very difficult to capture in the act of migrating through the basement membrane [41], it seems unlikely that if this actually occurred, after decades of modern pathology a renal pathologist would not have made this observation by now.
In parallel with the studies summarized above that have questioned the existence of EMT as a source of myofibroblasts in renal fibrosis, a similar re-evaluation of the existence of EMT as a source of myofibroblasts in liver fibrosis is underway. Using genetic fate-mapping approaches, three independent groups have recently found no evidence that hepatocytes undergo EMT to become collagen-producing myofibroblasts [37, 47, 48], as had previously been thought [49, 50].
Other sources of myofibroblasts: Bone marrow and endothelium
The current literature suggests that bone-marrow-derived cells may also, to a very small extent, contribute to the population of matrix-producing cells in tissue fibrosis, showing parallels to Cohnheim’s original description almost a century and a half ago [51]. These cells are commonly referred to as “fibrocytes”—marrow-derived and blood-borne cells capable of leaving the blood, penetrating into tissue, and becoming fibroblasts. They share markers of both leukocytes and mesenchymal cells and their estimated contribution to the myofibroblast population appears to vary from organ to organ. In kidney, they were reported to account for ∼12% of tubulointerstitial fibroblasts in normal rodent kidney and proportionally somewhat more in fibrosis (∼15%) [22]. Those studies relied on the promoter for S100A4/FSP-1 to detect such cells, but the S100A4/FSP-1 gene is expressed in many cells, including macrophages, suggesting that those numbers may have been an over-estimate. In fact, more recently the notion that fibrocytes contribute substantially to kidney pathology has been challenged using collagen 1 promoter activity to report fibrocytes. Those studies failed to detect a significant contribution of circulating cells to renal myofibroblasts in mouse kidney [26, 52].
Very recently, it has been reported that endothelial cells may represent yet another progenitor pool for fibroblasts and myofibroblasts. In an article published in 2008, Zeisberg and colleagues argue that fibroblasts in kidney fibrosis emerge via a process termed endothelial-to-mesenchymal transition [53]. This conclusion was based on their observation that 30 to 50% of fibroblasts coexpressed the endothelial marker CD31 and markers of fibroblasts and myofibroblasts such as FSP-1 and αSMA as analyzed by immunostaining in three different models of kidney fibrosis. Using lineage-tracing techniques crossing a Tie2-Cre mouse as “endothelial specific” Cre-driver with a Rosa26-stop-EYFP reporter mouse they further confirm the presence of EndMT-derived fibroblasts in fibrotic kidney. These observations bear analogy to findings in cardiac fibrosis, in which EndMT was first proposed as an important mechanism of fibrogenesis by the same group using a similar approach [54]. These results will require confirmation using other endothelial-specific Cre drivers.
Pericyte or resident fibroblast?
Despite much controversy and debate, arguably the most “traditional” concept, which holds that matrix-producing cells derive from local stromal cells, is still accepted by many as the main route of myofibroblast recruitment [55]. In an attempt to shed light on this putative myofibroblast progenitor pool, we genetically labeled kidney stromal cells using the FoxD1-Cre driver mouse and crossing it to various reporter lines (Fig. 2). FoxD1 (forkhead box D1) is a transcription factor that is prominently expressed during nephrogenesis in cells surrounding the cap condensate fated to become resident perivascular cells, pericytes, vascular smooth muscle and mesangial cells [56, 57]. Healthy kidneys from FoxD1-Cre; R26R mice exhibited strong β-gal (product of LacZ) expression in cells located in the tubulointerstitium of cortex and medulla [32]. Immunostaining found these cells to be F4/80-, CD31-, αSMA- but CD73+ and PDGF-Rβ+. Following UUO injury, this population showed marked expansion and gain of αSMA expression, consistent with the notion that FoxD1-labeled progenitors converted into myofibroblasts. In fact, these cells accounted for the vast majority of myofibroblasts in the diseased kidney, thus justifying a redirection of focus and renewed emphasis on local stromal cells as actual progenitor pool.

Genetically labeled pericytes in adult kidney. FoxD1-Cre driver was crossed to R26-tdtomato reporter mice resulting in recombination and permanent labeling (red) of renal stromal cells, including pericytes and perivascular fibroblasts, in bigenic offspring. Note the delicate spindle shape and long processes extending around the tubules that characterize labeled cells in the renal interstitium. Nuclei were counterstained with DAPI and the image was captured by confocal microscopy at 400x magnification
The nomenclature of Foxd1-derived stromal cells in the adult kidney interstitium is currently a matter of debate. While some of the controversy stems from differences in historic documentations and the particular context in which these cells were first described, others seem to be of mere semantic nature. For instance, pericytes—also known as mural or “Rouget” cells—have until recently only been referred to in a stringently anatomic or etymologic sense, that is ‘peri’-endothelial cells surrounding capillaries and venules, without any functional connotation. Initially described by French scientist Charles-Marie Benjamin Rouget almost 140 years ago [58], pericytes were first unequivocally identified in the kidney in 1983 using electron microscopy [59]. They are an integral part of peritubular capillaries and postcapillary venules. Kidney pericytes as described by Courtnoy and Boyles are sheathed, fully or in part, with basement membrane and make close contacts with endothelium, thus fulfilling the major criteria of pericytes. The sheathing is thought to be a duplication of the capillary basement membrane (CBM), which is often found to be incomplete between pericyte and endothelial cells, hereby enabling close apposition and interdigitation between both cell types to occur (Fig. 3). These CBM “gaps” frequently coincide with adhesion plaques and variants of pericyte projections, which are believed to be sites of cell–cell signaling. In kidney, some pericytes span from the peritubular capillary to the tubule with processes abutting the tubular basement membrane and may be capable of bilateral signaling—with endothelium as well as epithelium. In this context, they have been called resident fibroblasts [60]. Surrounding arterioles and larger vessels, these cells adhere to the vessel wall but are not in direct communication with the endothelium. Here they are often referred to as perivascular fibroblasts and adventitial cells, respectively.

EM photomicrographs of peritubular capillaries from human kidney. a Micrograph demonstrating the relationship of pericytes to endothelial cells. Of note, the pericyte is sheathed by duplication of the capillary basement membrane (CBM capillary basement membrane; cf collagen fibers; EC endothelial cell; Mϕ resident macrophage; P pericyte; Pp pericyte process; PTC peritubular capillary; RBC red blood cell; Tu tubule). b A pericyte with characteristic projections abutting and within the CBM. c Extensive pericyte process within cortical CBM. Insert shows higher power image of the tip of the process, and arrowhead indicates the reflection of the CBM and junction with the endothelial cell
We suggest that the majority of FoxD1-labeled interstitial cells are indeed true pericytes, and not simply fibroblasts. Pericytes are defined anatomically by their close apposition to microvessels with extensively branched processes that make close contacts to endothelium [61]. Such cells are clearly present in kidney interstitium (Fig. 3, [59]). Whether all FoxD1-labeled interstitial cells make these close contacts to endothelium is currently unresolved, but given the highly branched nature of these cells (Fig. 2) and the high density of the capillary network surrounding tubules, this possibility cannot be excluded. The absence of αSMA expression in these kidney cells has been suggested as another reason that these cells may not be properly termed ‘pericytes’, since contractile function and αSMA expression typically coincide in pre- and postcapillary pericytes in other vascular beds. However, mid-capillary pericytes have been shown to lack αSMA expression, for example in mesenteric mid-capillaries and retinal capillaries in rat [62], and mid-capillary pericytes from human breast [63]. Moreover, the absence of αSMA expression in kidney pericytes does not imply that these cells cannot play a role in regulating local blood flow, as pericytes in other organs do, since they may express other contractile machinery or control blood flow by paracrine mechanisms.
Emerging data suggests that this cell compartment may have more heterogeneity and unrecognized potential than previously appreciated. Pericytes in other tissue beds are necessary for sprouting angiogenesis and vascular stabilization, not only developmentally but also in repair following injury. Intriguingly, recent studies have indicated the existence of similarities between pericytes and mesenchymal stem cells (MSC) [64]. Studies are underway to dissect whether kidney pericytes serve similar functions in the kidney. In models of fibrotic kidney disease, we and others have observed that these cells can respond to chronic injury and profibrotic signals by proliferation, migration, and scar tissue formation, resulting in early perivascular and later diffuse renal fibrosis [26, 32, 43–45] (Fig. 4). In a very recent follow-up study, we could show that pericyte migration away from vasculature is in fact a central feature of fibrotic kidney disease that contributes to microvascular rarefaction, and that targeting communication between pericytes and endothelium may constitute a useful anti-fibrotic strategy [65]. This study emphasizes the important crosstalk between kidney pericytes and vasculature and highlights a vascular-stabilizing role for kidney pericytes.

Myofibroblasts originating from kidney pericytes and perivascular fibroblasts. The schematic shows the perception of myofibroblast recruitment from local pericytes and perivascular fibroblasts in the setting of chronic kidney injury (↓). Persistent irritation and injury lead to activation and transformation of pericytes and perivascular fibroblasts into myofibroblasts with subsequent migration, proliferation, and deposition of extracellular matrix (ECM) in tubulointerstitial spaces. The consequences are progressive scar tissue formation and loss of functional renal parenchyma
Future questions
There is now sufficient evidence to demand a critical reassessment of the paradigm that epithelial cells undergo full transformation into myofibroblasts and migrate into interstitium in CKD, but the question remains of what exact role injury to tubule epithelium (some may choose to call this partial mesenchymal transition) may play in regulation of fibrosis. Paracrine signaling of this ‘intermediate phenotype’, for instance, has been proposed as an important contributor to fibrogenesis [36, 66], but the signaling pathways and transcriptional circuitries that govern epithelial dedifferentiation and whether manipulation of these pathways could be exploited for therapeutic purposes, are largely unknown.
Other unanswered questions include, what are the consequences of pericyte/perivascular fibroblast migration and what effect does recruitment to the myofibroblast pool have on the homeostasis of renal microvasculature? What heterogeneity exists among pericytes, perivascular fibroblasts, and adventitial cells, and are certain pericyte subgroups more or less likely to be recruited into the myofibroblast pool? If so, what markers distinguish such pericyte subsets and will they allow more accurate targeting of the myofibroblast progenitor pool? Efforts to dissect and understand these biological properties will clearly help in identifying new therapeutic targets and in developing novel anti-fibrotic strategies. Finally, especially in view of indications that pericytes may represent a tissue-specific MSC niche, it will be important to purify and enrich these cells, develop culture conditions, and investigate their multi-potentiality.
Summary
Understanding the origins of myofibroblasts in renal fibrosis is critical for targeting anti-fibrotic strategies to the correct precursor population. Recent genetic lineage analysis suggests that contrary to widely accepted theories, epithelial cells do not become myofibroblasts during EMT. Rather, pericytes and perivascular fibroblasts differentiate into myofibroblasts, the matrix-secreting cell type in chronic fibrotic kidney disease. Defining the pathways that regulate activation of these myofibroblast precursors after injury is a promising future research area for the identification of novel CKD biomarkers and therapeutic targets.
References
Wynn TA (2007) Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest 117:524–529
Wynn TA (2008) Cellular and molecular mechanisms of fibrosis. J Pathol 214:199–210
Bohle A, Strutz F, Muller GA (1994) On the pathogenesis of chronic renal failure in primary glomerulopathies: a view from the interstitium. Exp Nephrol 2:205–210
Nankivell BJ, Fenton-Lee CA, Kuypers DR, Cheung E, Allen RD, O'Connell PJ, Chapman JR (2001) Effect of histological damage on long-term kidney transplant outcome. Transplantation 71:515–523
Qi W, Chen X, Poronnik P, Pollock CA (2006) The renal cortical fibroblast in renal tubulointerstitial fibrosis. Int J Biochem Cell Biol 38:1–5
Skalli O, Ropraz P, Trzeciak A, Benzonana G, Gillessen D, Gabbiani G (1986) A monoclonal antibody against alpha-smooth muscle actin: a new probe for smooth muscle differentiation. J Cell Biol 103:2787–2796
Serini G, Gabbiani G (1999) Mechanisms of myofibroblast activity and phenotypic modulation. Exp Cell Res 250:273–283
Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA (2002) Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 3:349–363
Darby I, Skalli O, Gabbiani G (1990) Alpha-smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab Invest 63:21–29
Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G (1993) Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol 122:103–111
Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, Gabbiani G (1998) The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol 142:873–881
Bechtel W, McGoohan S, Zeisberg EM, Muller GA, Kalbacher H, Salant DJ, Muller CA, Kalluri R, Zeisberg M (2010) Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med 16:544–550
Cohnheim J (1867) Ueber Entzuendung und Eiterung. Virchows Arch 40:1–79
Ross R, Everett NB, Tyler R (1970) Wound healing and collagen formation. VI. The origin of the wound fibroblast studied in parabiosis. J Cell Biol 44:645–654
Kalluri R, Neilson EG (2003) Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest 112:1776–1784
Liu Y (2010) New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol 21:212–222
Greenburg G, Hay ED (1982) Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J Cell Biol 95:333–339
Thiery JP, Acloque H, Huang RY, Nieto MA (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139:871–890
Yang J, Weinberg RA (2008) Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 14:818–829
Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Invest 119:1420–1428
Liu Y (2004) Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol 15:1–12
Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG (2002) Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 110:341–350
Witzgall R, Brown D, Schwarz C, Bonventre JV (1994) Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J Clin Invest 93:2175–2188
Grone HJ, Weber K, Grone E, Helmchen U, Osborn M (1987) Coexpression of keratin and vimentin in damaged and regenerating tubular epithelia of the kidney. Am J Pathol 129:1–8
Le Hir M, Hegyi I, Cueni-Loffing D, Loffing J, Kaissling B (2005) Characterization of renal interstitial fibroblast-specific protein 1/S100A4-positive cells in healthy and inflamed rodent kidneys. Histochem Cell Biol 123:335–346
Lin SL, Kisseleva T, Brenner DA, Duffield JS (2008) Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol 173:1617–1627
Li L, Zepeda-Orozco D, Black R, Lin F (2010) Autophagy is a component of epithelial cell fate in obstructive uropathy. Am J Pathol 176:1767–1778
Joyner AL, Zervas M (2006) Genetic inducible fate mapping in mouse: establishing genetic lineages and defining genetic neuroanatomy in the nervous system. Dev Dyn 235:2376–2385
Duffield JS, Humphreys BD (2010) Origin of new cells in the adult kidney: results from genetic labeling techniques. Kidney Int. doi:10.1038/ki.2010.338
Hayashi S, McMahon AP (2002) Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 244:305–318
Humphreys BD, Valerius MT, Kobayashi A, Mugford JW, Soeung S, Duffield JS, McMahon AP, Bonventre JV (2008) Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2:284–291
Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS (2010) Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176:85–97
Endo T, Okuda T, Nakamura J, Higashi AY, Fukatsu A, Kita T, Yanagita M (2010) Exploring the origin of the cells responsible for regeneration and fibrosis in the kidneys. J Am Soc Nephrol (Abstract) 21:F-FC163.
Koesters R, Kaissling B, Lehir M, Picard N, Theilig F, Gebhardt R, Glick AB, Hahnel B, Hosser H, Grone HJ, Kriz W (2010) Tubular overexpression of transforming growth factor-beta1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol 177:632–643
Zeisberg M, Bottiglio C, Kumar N, Maeshima Y, Strutz F, Muller GA, Kalluri R (2003) Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am J Physiol Renal Physiol 285:F1060–1067
Bielesz B, Sirin Y, Si H, Niranjan T, Gruenwald A, Ahn S, Kato H, Pullman J, Gessler M, Haase VH, Susztak K (2010) Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest 120:4040–4054
Taura K, Miura K, Iwaisako K, Osterreicher CH, Kodama Y, Penz-Osterreicher M, Brenner DA (2010) Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology 51:1027–1036
Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, Pickel J, McKay R, Nadal-Ginard B, Bodine DM, Leri A, Anversa P (2001) Bone marrow cells regenerate infarcted myocardium. Nature 410:701–705
Murry CE, Soonpaa MH, Reinecke H, Nakajima H, Nakajima HO, Rubart M, Pasumarthi KB, Virag JI, Bartelmez SH, Poppa V, Bradford G, Dowell JD, Williams DA, Field LJ (2004) Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature 428:664–668
Balsam LB, Wagers AJ, Christensen JL, Kofidis T, Weissman IL, Robbins RC (2004) Haematopoietic stem cells adopt mature haematopoietic fates in ischaemic myocardium. Nature 428:668–673
Zeisberg M, Duffield JS (2010) Resolved: EMT produces fibroblasts in the kidney. J Am Soc Nephrol 21:1247–1253
Cook HT (2010) The origin of renal fibroblasts and progression of kidney disease. Am J Pathol 176:22–24
Wiggins R, Goyal M, Merritt S, Killen PD (1993) Vascular adventitial cell expression of collagen I messenger ribonucleic acid in anti-glomerular basement membrane antibody-induced crescentic nephritis in the rabbit. A cellular source for interstitial collagen synthesis in inflammatory renal disease. Lab Invest 68:557–565
Faulkner JL, Szcykalski LM, Springer F, Barnes JL (2005) Origin of interstitial fibroblasts in an accelerated model of angiotensin II-induced renal fibrosis. Am J Pathol 167:1193–1205
Picard N, Baum O, Vogetseder A, Kaissling B, Le Hir M (2008) Origin of renal myofibroblasts in the model of unilateral ureter obstruction in the rat. Histochem Cell Biol 130:141–155
Grone HJ, Kriz W (2010) Cells involved in renal tubulointerstitial degeneration and fibrosis—A matter of differentiation or cell lineage switch? ISN nexus Fibrosis and the Kidney. International Society of Nephrology, Geneva, Switzerland.
Scholten D, Osterreicher CH, Scholten A, Iwaisako K, Gu G, Brenner DA, Kisseleva T (2010) Genetic labeling does not detect epithelial-to-mesenchymal transition of cholangiocytes in liver fibrosis in mice. Gastroenterology 139:987–998
Duffield JS (2010) Epithelial to mesenchymal transition in injury of solid organs: fact or artifact? Gastroenterology 139(4):1081–1083
Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, Kalluri R (2007) Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem 282:23337–23347
Wells RG (2010) The epithelial-to-mesenchymal transition in liver fibrosis: here today, gone tomorrow? Hepatology 51:737–740
Wada T, Sakai N, Matsushima K, Kaneko S (2007) Fibrocytes: a new insight into kidney fibrosis. Kidney Int 72:269–273
Roufosse C, Bou-Gharios G, Prodromidi E, Alexakis C, Jeffery R, Khan S, Otto WR, Alter J, Poulsom R, Cook HT (2006) Bone marrow-derived cells do not contribute significantly to collagen I synthesis in a murine model of renal fibrosis. J Am Soc Nephrol 17:775–782
Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R (2008) Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol 19:2282–2287
Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R (2007) Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 13:952–961
Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G (2007) The myofibroblast: one function, multiple origins. Am J Pathol 170:1807–1816
Levinson RS, Batourina E, Choi C, Vorontchikhina M, Kitajewski J, Mendelsohn CL (2005) Foxd1-dependent signals control cellularity in the renal capsule, a structure required for normal renal development. Development 132:529–539
Hatini V, Huh SO, Herzlinger D, Soares VC, Lai E (1996) Essential role of stromal mesenchyme in kidney morphogenesis revealed by targeted disruption of Winged Helix transcription factor BF-2. Genes Dev 10:1467–1478
Rouget C (1873) Memoire sur le developpement, la structure et les proprietes physiologiques des capillaires sanguins et lymphatiques. Arch Physiol Norm et Path 5:603–663
Courtnoy P, Boyles J (1983) Fibronectin in the microvasculature: localization in the pericyte-endothelial interstitium. J Ultrastruct Res 83:258–273
Kaissling B, Hegyi I, Loffing J, Le Hir M (1996) Morphology of interstitial cells in the healthy kidney. Anat Embryol (Berl) 193:303–318
Diaz-Flores L, Gutierrez R, Madrid JF, Varela H, Valladares F, Acosta E, Martin-Vasallo P, Diaz-Flores L Jr (2009) Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol Histopathol 24:909–969
Nehls V, Drenckhahn D (1991) Heterogeneity of microvascular pericytes for smooth muscle type alpha-actin. J Cell Biol 113:147–154
Ronnov-Jessen L, Petersen OW, Koteliansky VE, Bissell MJ (1995) The origin of the myofibroblasts in breast cancer. Recapitulation of tumor environment in culture unravels diversity and implicates converted fibroblasts and recruited smooth muscle cells. J Clin Invest 95:859–873
Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, Andriolo G, Sun B, Zheng B, Zhang L, Norotte C, Teng PN, Traas J, Schugar R, Deasy BM, Badylak S, Buhring HJ, Giacobino JP, Lazzari L, Huard J, Peault B (2008) A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 3:301–313
Lin S, Chang F, Schrimpf C, Chen Y, Wu C, Wu V, Chiang W, Kuhnert F, Kuo CJ, Chen Y, Wu K, Tsai T, Duffield JS (2011) Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am J Pathol 178:911–923
Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV (2010) Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16:535–543, 531p following 143
Acknowledgements
The authors wish to thank Dr. Vanesa Bijol and Colleen Ford (Renal Pathology, Brigham and Women’s Hospital) for generous contribution of EM micrographs and helpful discussions. I.G. is supported by a fellowship from the Deutsche Forschungsgemeinschaft (GR 3301/4-1) and grants from the German Kidney Foundation, German Society of Hypertension, and University Medical Center Giessen and Marburg. Laboratory of Inflammation Research is supported by DK73299, DK84077, DK87389, and the Institute for Stem Cell & Regenerative Medicine, Seattle, WA. Work in the Humphreys Laboratory is supported by NIH grants DK88923, DK84316, and DK73628 and funds from the Harvard Stem Cell Institute.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Grgic, I., Duffield, J.S. & Humphreys, B.D. The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr Nephrol 27, 183–193 (2012). https://doi.org/10.1007/s00467-011-1772-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-011-1772-6