
Abstract
Fibroblast growth factor receptors (Fgfrs) are expressed throughout the developing kidney. Several early studies have shown that exogenous fibroblast growth factors (Fgfs) affect growth and maturation of the metanephric mesenchyme (MM) and ureteric bud (UB). Transgenic mice that over-express a dominant negative receptor isoform develop renal aplasia/severe dysplasia, confirming the importance of Fgfrs in renal development. Furthermore, global deletion of Fgf7, Fgf10, and Fgfr2IIIb (isoform that binds Fgf7 and Fgf10) in mice leads to small kidneys with fewer collecting ducts and nephrons. Deletion of Fgfrl1, a receptor lacking intracellular signaling domains, causes severe renal dysgenesis. Conditional targeting of Fgf8 from the MM interrupts nephron formation. Deletion of Fgfr2 from the UB results in severe ureteric branching and stromal mesenchymal defects, although loss of Frs2α (major signaling adapter for Fgfrs) in the UB causes only mild renal hypoplasia. Deletion of both Fgfr1 and Fgfr2 in the MM results in renal aplasia with defects in MM formation and initial UB elongation and branching. Loss of Fgfr2 in the MM leads to many renal and urinary tract anomalies as well as vesicoureteral reflux. Thus, Fgfr signaling is critical for patterning of virtually all renal lineages at early and later stages of development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Congenital kidney diseases are the leading causes of chronic kidney disease in children [1]. Although the underlying genetic defects leading to structural kidney disease are largely unknown, there are syndromes in which mutations in fibroblast growth factor receptors (FGFRs) lead to renal anomalies. For example, activating mutations of FGFR1 and/or 2 cause syndromes, such as Apert’s syndrome, Antley-Bixler syndrome, Pfeiffer syndrome, and Beare-Stevenson syndrome, which are sometimes associated with urogenital anomalies such as hydroureter, solitary kidney, and VUR [2–5]. Loss-of-function mutations in FGFR1 have also been associated with some variants of Kallman syndrome that can be associated with unilateral renal aplasia. Rarely, activating mutations in FGFR3 leading to thanatophoric dysplasia are associated with renal hypoplasia or cystic dysplasia [6, 7]. Given recent technological advances in genome and exome sequencing, it is possible that many of the sporadic cases of congenital renal anomalies will be associated with polymorphisms or copy number variations in FGFRs. The remainder of this review will focus on tissue and animal studies that are beginning to clarify the role of Fgfr signaling in kidney development.
Background on kidney development and fibroblast growth factor receptors
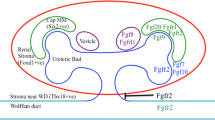
The metanephric kidney develops from tissues primarily arising from the intermediate mesoderm, the Wolffian (nephric) duct, and the nephrogenic cord [8]. Between embryonic days (E) 10.5-11.0 in the mouse, the nephrogenic cord gives rise to the metanephric mesenchyme (MM), which then induces the formation of the ureteric bud (UB) from the Wolffian duct in the region of the hindlimb [8]. Stromal mesenchyme lying between the Wolffian duct and the MM restricts the UB to its proper position and prevents ectopic budding [9]. As the ureteric bud elongates, the metanephric mesenchyme divides into a nephrogenic lineage lying adjacent to the bud and a surrounding renal cortical stromal lineage (that may also have elements of paraxial mesoderm) [8, 10]. The kidney continues to develop due to reciprocal inductive interactions with the ureteric bud receiving signals to branch dichotomously, ultimately to form the collecting ducts and ureters [8]. At each terminal tip, the ureteric bud induces local areas of nephrogenic mesenchyme to differentiate into nephron epithelia, progressing from renal vesicles, to comma-shaped bodies, to S-shaped bodies, and finally to mature nephrons [8].
Fibroblast growth factor receptors (Fgfrs) are receptor tyrosine kinases with four signaling family members that are activated by binding to one of 22 known Fgf ligands in mammals [11]. Fgfr proteins consist of up to three external immunoglobulin (Ig)-like domains, a transmembrane domain, and an intracellular kinase domain [11]. Fgfrs1-3 exist as either a IIIb or IIIc isoform, through alternate splicing of the C-terminal portion of the third Ig-like domain, whereas Fgfr4 only has a IIIc isoform [11]. These splice variants have different ligand-binding specificities; for instance, Fgf7 and Fgf10 both bind to Fgfr2IIIb but not to Fgfr2IIIc [11]. In addition, IIIb isoforms are usually expressed in epithelium, while IIIc isoforms are present in mesenchyme [11]. All of the receptor isoforms utilize molecules, such as fibroblast growth factor receptor substrate 2α (Frs2α), and phospholipase Cγ (PLCγ), to transmit intra-cellular signaling [11]. Frs2α constitutively binds Fgfrs in the juxtamembrane region and upon receptor activation becomes phosphorylated, then activating Erk, Akt, and protein kinase C (PKC) λ and ξ. PLCγ is recruited to a phosphorylated tyrosine on activated receptors, leading to activation of PKC (and possibly Src and Grb2). Given that all of the receptor isoforms use the same intracellular signaling molecules, much of the specificity of the pathway is likely dictated by the ligands. In addition to the four signaling Fgfrs, fibroblast growth factor receptor-like 1 (Fgfrl1, formerly Fgfr5) is a molecule with an extracellular domain highly homologous to Fgfrs that can bind Fgf ligands, but it lacks a tyrosine kinase domain for intracellular signaling [12].
Although Fgfr3 and Fgfr4 have been detected in embryonic kidneys [13–15], Fgfr1, Fgfr2, and Fgfrl1 appear to be the most relevant receptors regarding renal development (see below). Fgfr1 is expressed most prominently in rodent mesenchymal tissues (early metanephric mesenchyme, condensing mesenchyme, and developing nephrons), but is present at lower levels in the ureteric lineage and in renal cortical stroma [16–20]. Fgfr2 is strongly expressed in the Wolffian duct and the ureteric bud tree (tips and trunks) and differentiating nephrons, but is present at lower levels in early MM and stromal mesenchyme adjacent to the Wolffian duct and main ureteric trunk [13, 16–20]. Fgfrl1 is present in renal vesicles [12].
Roles of Fgfs in regulating renal development
The first studies suggesting relevance of Fgfr signaling in renal development focused on the addition of Fgf ligands to isolated embryonic kidney tissues or on overexpression of ligands in vivo. In rodent and Xenopus explants, exogenous Fgf2 (alone or in combination with other growth factors) was able to sustain mesenchymal tissue growth and in some cases to induce formation of mature nephrons [21–25]. In addition, Fgf1, Fgf2, Fgf7, and Fgf10 stimulated growth and differentially affected branching morphogenesis in isolated rat ureteric bud cultures and/or in intact rat embryonic kidney explants [26, 27]. Finally, overexpression of FGF2 (basic fibroblast growth factor) or FGF7 (keratinocyte growth factor) in developing rodent kidneys in vivo, leads to cystic dilation of collecting ducts [28, 29]. Thus, the addition or overexpression of fibroblast growth factors in vitro and in vivo affects growth and maturation of both mesenchymal and ureteric bud lineages in the developing kidney.
Mouse knockout studies have verified the relevance of Fgf signaling in the embryonic kidney (Table 1). Targeted deletion of Fgf7 (produced by cortical stromal cells) leads to viable mice with a reduction in ureteric branch number and fewer nephrons, likely due to decreased signaling through Fgfr2IIIb receptors on the ureteric epithelium [27]. Deletion Fgf10, another ligand expressed in metanephric mesenchyme with specificity for Fgfr2IIIb, resulted in perinatal lethality likely from severe dysgenesis/agenesis of lungs and limbs; similar to Fgf7-/- mice, Fgf10-/- mice had smaller kidneys and fewer collecting ducts [30]. Fgf8, expressed in early MM condensates, is an attractive candidate ligand mediating nephron development; however, Fgf8-/- mice are early embryonic lethal [31]. To circumvent the early lethality of Fgf8 null mice, two groups of investigators used a conditional knockout approach with Pax3creTg/+ transgenic mice (that express cre recombinase in the MM) or TcreTg/+ transgenic mice (that express cre in all mesodermal tissues) [32, 33]. The resulting mice that had no functional Fgf8 in the metanephric mesenchyme (Fgf8Mes-/-) survived until birth, but had small kidneys with an interruption in nephron formation after the epithelial vesicle stage. Thus, Fgfr signaling appears crucial for ureteric branching morphogenesis and nephron maturation.
Effects of genetic blockade of Fgfr signaling on the developing kidney
The first mouse with altered Fgfr expression leading to renal abnormalities was a transgenic line carrying a solubilized Fgfr2IIIb fragment, which acted as a dominant negative by binding many different Fgfs (and thus preventing much of the signaling through endogenous Fgfrs) [34]. The mutant embryos developed severe renal dysgenesis or agenesis (as well as severe lung, exocrine and endocrine gland, cutaneous, and limb anomalies) [34]. Regarding specific receptors, mice with deletion of Fgfr3 or Fgfr4 alone or in combination are viable and have no obvious kidney abnormalities [35, 36, and unpublished data], making these receptors unlikely candidates to mediate signaling in the developing kidney. Global targeting of Fgfr2IIIb (the receptor isoform for both Fgf7 and Fgf10) results in a renal phenotype similar to the Fgf7 and Fgf10 knockouts, including smaller kidneys with fewer nephrons than normal [37] (Table 1). Deletion of Fgfrl1 resulted in severe renal dysgenesis secondary to interruption of nephron differentiation and ureteric branching [12] (Table 1). More recently, Fgfrl1 was shown to act in part as a decoy receptor, competing for ligands with signaling Fgfrs [38]; thus, a proper balance of Fgfr signaling may be crucial for normal metanephric development.
Roles of Fgfr1 and Fgfr2 (outside of Fgfr2IIIb) in the kidney have been difficult to ascertain with global knockout techniques due to early embryonic lethality of Fgfr1 and Fgfr2 null mice [39–42]. To circumvent the early lethality of Fgfr1 and Fgfr2 knockout mice, our laboratory used conditional knockout strategies in different renal lineages (Table 1). We first bred Hoxb7creEGFPTg/+ transgenic mice (expressing cre recombinase and a green fluorescent protein in the Wolffian duct and ureteric lineage) to floxed Fgfr mice to delete the receptors in the ureteric lineage (FgfrUB-/-) [18]. While all mice were viable, Fgfr2UB-/- mice had small kidneys with ureteric branching defects, including longer branches and fewer tips (total and corrected for surface area) (Fig. 1). Fgfr1UB-/- mice had no defects and Fgfr1/2UB-/- were indistinguishable from Fgfr2UB-/-. Fgfr2UB-/- mice also had thickened cortical stroma with no interdigitations and many fewer nephrons than controls, likely from fewer ureteric tips.

Comparison of control and Fgfr2UB-/- ureteric bud branching in explants and by 3D reconstructive imaging. Fluorescent micrographs of E11.5 explants after 3 days of growth demonstrating that compared with controls (a) Fgfr2UB-/- explants (b) have thin, long ureteric trunks and fewer ureteric bud tips with thin ampullae (40x magnification). 3D reconstructive imaging of E13.5 kidneys confirm that compared with controls (c), Fgfr2UB-/- mutant (d) ureteric epithelium occupies less relative volume and has fewer but longer ureteric segments. Scale bar = 100μm. (Con control, Mut mutant) (a, b reprinted from Fig. 5 in [18] with permission from Elsevier, and c and d reprinted from Fig. 2 in [43] with permission from the American Society of Nephrology)
We recently characterized Fgfr2UB-/- kidneys by three-dimensional (3D) reconstructive imaging that revealed more striking defects and allowed for better quantification of abnormalities than in explants [43] (Fig. 1). In the ureteric lineage, mutants had more dramatic decreases in tip number versus controls (59% 3D vs. 42% in explants). 3D reconstructions also allowed quantification of a reduction in Fgfr2UB-/- ureteric tissue per kidney volume and increases in mutant median ureteric segment lengths versus the controls. In addition to revealing striking decreases in Fgfr2UB-/- nephron number (vs. controls), 3D reconstructions also revealed that mean Fgfr2UB-/- developing nephron sizes were larger than the controls; mutant vesicles are the same size as controls but gradually increase in volume and surface area, reaching statistical differences as mature glomeruli. Since murine nephrons are not yet filtering urine at E13.5, the increase in glomerular size is not likely the result of “compensatory hypertrophy”. Thus, Fgfr2 in the ureteric lineage is critical for normal ureteric branching and as a secondary consequence, nephron development.
To determine the role of Frs2α, a major intracellular signaling adapter for Fgfrs, in the ureteric lineage, we generated conditional knockout mice using the Hoxb7cre line (Frs2αUB-/- [44]. Surprisingly, Frs2αUB-/- mice developed only mild renal hypoplasia characterized by decreased ureteric branching events, but with normal overall branching architecture and normal stromal mesenchymal development. The alterations in ureteric morphogenesis were likely secondary to decreased Ret and Wnt11 expression characterized by in situ hybridization and real-time PCR. There were also minor reductions in nephron endowment secondary to the decrease in ureteric tip number. Thus Frs2αUB-/- mice had a much more mild renal phenotype than Fgfr2UB-/- mice, suggesting that Fgfr2 likely signals through other adapter molecules in the ureteric epithelium. In addition to Fgfrs, Frs2α transmits intracellular signaling for other receptor tyrosine kinases such as Ret, neurotrophin receptors, and anaplastic lymphoma kinase [45].
To determine whether Fgfr2 signals through Frs2α at all in the ureteric epithelium, we generated mice with point mutations in the Frs2α binding site on Fgfr2 (termed Fgfr2LR based on the amino acids that were mutated to alanine—such point mutations have been shown to abrogate Frs2α-Fgfr2 binding, while not affecting signaling through other adapter proteins) [46]. Fgfr2LR/LR mice had phenotypically normal kidneys based on histology, explant analysis, and size measurements [44]. To avoid any potential rescue by intact Fgfr1 forming heterodimers with Fgfr2LR, we are currently examining mice that are Hoxb7creTg/+Fgfr1Lox/LoxFgfr2LR/LR compound mutant mice. If the compound mutant mice are phenotypically normal, it would suggest that Frs2α serves primarily as an adapter protein for other receptor tyrosine kinases in the ureteric epithelium, such as Ret.
To investigate the role(s) of Fgfrs in MM, we used conditional knockout approaches with the Pax3creTg/+ transgenic mouse line (FgfrMes-/-) [19]. While deletion of any three out of four Fgfr1 and Fgfr2 alleles generally resulted in robust kidney formation, deletion of all four alleles (Fgfr1/2Mes-/-) resulted in severe renal dysgenesis with no recognizable MM at E10.5 (Fig. 2). MM markers, Eya1 and Six1, were actually present in a very restricted domain, but downstream markers Six2, Sall1, and Pax2 were absent in mutant kidney mesenchyme. In the ureteric lineage, E10.5 Fgfr1/2Mes-/- embryos had ureteric outgrowth (and occasionally multiple buds) secondary to glial cell line-derived neurotrophic factor (Gdnf) expression in the MM; however, by E11.5 Gdnf was no longer present, leading to no ureteric elongation or branching. Thus, Fgfr1 and Fgfr2 signaling (together) in the metanephric mesenchyme are critical for formation of the metanephric mesenchyme and early development of the ureteric bud. To determine whether the early MM patterning defects seen in Fgfr1/2Mes-/- are dependent on Frs2α-mediated signaling, we are currently examining compound mutant mice with Pax3cre-mediated deletion of Fgfr1 and with Fgfr2LR point mutations.

Comparison of the E10.5 metanephric mesenchyme and ureteric bud in control and Fgfr1/2Mes-/- mice. Hematoxylin & eosin stains of transverse sections through caudal intermediate mesoderm demonstrate that control embryos (a) possess initial ureteric buds (arrowheads) and compacted metanephric mesenchymal (mm) tissues; Fgfr1/2Mes-/- mice (b) appear to possess initial ureteric buds (arrowheads) but no obvious metanephric mesenchyme (200x magnification) (Con control; Mut mutant) (Reprinted from Fig. 7 in [19] with permission from Elsevier)
Since Fgfr1/2Mes-/- mice sometimes had multiple UBs per Wolffian duct, we explored whether mice with single Pax3cre-driven Fgfr deletions had ureteric induction defects [47]. While Fgfr1Mes-/- were normal, 67% of E10.5-11.5 Fgfr2Mes-/- mice had two UBs per nephric duct (that often later fused into one UB). The UB induction defects resulted in structural defects in ∼25% of older embryos including duplex kidneys/collecting systems (partial and complete), aplasia, and obstructive hydroureter [47]. Key molecules known to regulate the UB induction site, including Gdnf, Robo2, Bone morphogenetic protein 4, and Sprouty1, were expressed normally by in situ hybridization; thus, mechanisms causing UB induction defects were unclear. Knowing that stromal mesenchyme around the nephric duct constrains the UB to its proper site, we examined expression of Fgfr2 in this region in mutants. Whole mount and section in situ hybridization revealed that Fgfr2 was deleted in these cells, possibly meaning that Fgfr2 acts in stromal tissue to constrain the UB induction site. To test this further, we have begun studies to conditionally delete Fgfr2 using a Tbx18creTg mouse line (gift from Fen Chen) [48] that drives cre expression in the stromal tissues but not the kidney mesenchyme.
Given that abnormally positioned ureteric buds are postulated to lead to vesicoureteral reflux (VUR) post-natally, we assessed whether Pax3cre-driven Fgfr2Mes-/- mutants had displaced ureteric buds and were predisposed to VUR. Compared to controls, Fgfr2Mes-/- embryos with single UBs per nephric duct had increased common nephric duct lengths indicating cranial displacement of the UBs along the Wolffian duct. Young Fgfr2 mutants also had high rates of VUR compared to controls (40% vs. 3.8%, p = 0.0003) (Fig. 3). 3D reconstructive imaging of mutants with unilateral reflux revealed that the refluxing ureters inserted much closer to the bladder neck than non-refluxing ureters (Fig. 3). Moreover, the external insertional angles of the ureter at the outer wall of the bladder (formed by the ureteral insertion points and bladder neck) were much greater in the mutant refluxing ureters compared to contralateral non-refluxing ureters as well as control ureters. Thus, Pax3cre-mediated deletion of Fgfr2 causes abnormal positioning of the ureteric bud, which is associated with abnormal ureteral insertion in the bladder and subsequent VUR. If Tbx18cre-mediated deletion of Fgfr2 results in ureteric induction abnormalities (see above), our laboratory will assess these mice for the presence of VUR as well.

Representative images cystograms and 3D reconstructions through the ureterovesical junction (UVJ) of newborn Fgfr2Mes-/- and control mice. Gravity-driven Methylene Blue cystograms show a control mouse (a) with no dye in either ureter (arrowheads) and a mutant mouse (b) with dye in the right ureter (arrowheads) and the pelvis of the right kidney (arrow) (12x magnification). c, d Posterior views of 3D reconstructions of the UVJ following cystograms show that insertion of the ureters (arrowheads) is at the same level in controls with no reflux (c); in the mutant (d) the right ureter (side of reflux, asterisk) inserts lower than the left (non-refluxing) ureter. Red tissue represents serosa and muscularis. Blue tissue represents ureteral urothelium. Scale bars = 100 μm. (Con control; Mut mutant) (R right side; L left side)
Conclusions
In conclusion, many fibroblast growth factors and all of their receptors are expressed in the developing kidney. In vitro and in vivo studies have shown that exogenous fibroblast growth factors affect both renal mesenchymal and ureteric bud development. Follow-up transgenic and gene knockout studies have shown that fibroblast growth factor receptor signaling is required for patterning of all renal lineages, including the ureteric bud, nephrogenic mesenchyme, and renal stromal mesenchyme. Furthermore, Fgfr activity is critical at early stages as well as later stages of kidney development. Also, there is clear redundancy between Fgfr1 and Fgfr2 in kidney mesenchyme. In humans, activating and loss-of-function mutations in FGFRs cause syndromes that are sometimes associated with urogenital anomalies [2–7]. Perturbations in fibroblast growth factor receptor signaling may also be responsible for sporadic cases of human renal congenital malformations and VUR.
References
(2010) North American Pediatric Renal Trials and Collaborative Studies: 2010 Annual Report. Rockville, pp 1–100
Passos-Bueno MR, Wilcox WR, Jabs EW, Sertie AL, Alonso LG, Kitoh H (1999) Clinical spectrum of fibroblast growth factor receptor mutations. Hum Mutat 14:115–125
Cohen MM Jr, Kreiborg S (1993) Visceral anomalies in the Apert syndrome. Am J Med Genet 45:758–760
Sergi C, Stein H, Heep JG, Otto HF (1997) A 19-week-old fetus with craniosynostosis, renal agenesis and gastroschisis: case report and differential diagnosis. Pathol Res Pract 193:579–585, discussion 587-578
Seyedzadeh A, Kompani F, Esmailie E, Samadzadeh S, Farshchi B (2008) High-grade vesicoureteral reflux in Pfeiffer syndrome. Urol J 5:200–202
Tohya T, Miura K, Nagata N (1986) A case of thanatophoric dwarfism with renal hypoplasia. Pediatr Int 28:232–237
Prontera P, Sensi A, Pilu G, Baldi M, Baffico M, Bonasoni R, Calzolari E (2006) FGFR3 mutation in thanatophoric dysplasia type 1 with bilateral cystic renal dysplasia: coincidence or a new association? Genet Couns 17:407–412
Dressler GR (2006) The cellular basis of kidney development. Annu Rev Cell Dev Biol 22:509–529
Miyazaki Y, Oshima K, Fogo A, Hogan BL, Ichikawa I (2000) Bone morphogenetic protein 4 regulates the budding site and elongation of the mouse ureter. J Clin Invest 105:863–873
Guillaume R, Bressan M, Herzlinger D (2009) Paraxial mesoderm contributes stromal cells to the developing kidney. Dev Biol 329:169–175
Powers CJ, McLeskey SW, Wellstein A (2000) Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer 7:165–197
Gerber SD, Steinberg F, Beyeler M, Villiger PM, Trueb B (2009) The murine Fgfrl1 receptor is essential for the development of the metanephric kidney. Dev Biol 335:106–119
Cancilla B, Ford-Perriss MD, Bertram JF (1999) Expression and localization of fibroblast growth factors and fibroblast growth factor receptors in the developing rat kidney. Kidney Int 56:2025–2039
Fuhrmann V, Kinkl N, Leveillard T, Sahel J, Hicks D (1999) Fibroblast growth factor receptor 4 (FGFR4) is expressed in adult rat and human retinal photoreceptors and neurons. J Mol Neurosci 13:187–197
Korhonen J, Partanen J, Alitalo K (1992) Expression of FGFR-4 mRNA in developing mouse tissues. Int J Dev Biol 36:323–329
Peters KG, Werner S, Chen G, Williams LT (1992) Two FGF receptor genes are differentially expressed in epithelial and mesenchymal tissues during limb formation and organogenesis in the mouse. Development (Cambridge, England) 114:233–243
Orr-Urtreger A, Givol D, Yayon A, Yarden Y, Lonai P (1991) Developmental expression of two murine fibroblast growth factor receptors, flg and bek. Development (Cambridge, England) 113:1419–1434
Zhao H, Kegg H, Grady S, Truong HT, Robinson ML, Baum M, Bates CM (2004) Role of fibroblast growth factor receptors 1 and 2 in the ureteric bud. Dev Biol 276:403–415
Poladia DP, Kish K, Kutay B, Hains D, Kegg H, Zhao H, Bates CM (2006) Role of fibroblast growth factor receptors 1 and 2 in the metanephric mesenchyme. Dev Biol 291:325–339
Dudley AT, Godin RE, Robertson EJ (1999) Interaction between FGF and BMP signaling pathways regulates development of metanephric mesenchyme. Genes Dev 13:1601–1613
Barasch J, Qiao J, McWilliams G, Chen D, Oliver JA, Herzlinger D (1997) Ureteric bud cells secrete multiple factors, including bFGF, which rescue renal progenitors from apoptosis. Am J Physiol 273:F757–F767
Perantoni AO, Dove LF, Karavanova I (1995) Basic fibroblast growth factor can mediate the early inductive events in renal development. Proc Natl Acad Sci USA 92:4696–4700
Barasch J, Yang J, Ware CB, Taga T, Yoshida K, Erdjument-Bromage H, Tempst P, Parravicini E, Malach S, Aranoff T, Oliver JA (1999) Mesenchymal to epithelial conversion in rat metanephros is induced by LIF. Cell 99:377–386
Plisov SY, Yoshino K, Dove LF, Higinbotham KG, Rubin JS, Perantoni AO (2001) TGF beta 2, LIF and FGF2 cooperate to induce nephrogenesis. Development (Cambridge, England) 128:1045–1057
Brennan HC, Nijjar S, Jones EA (1999) The specification and growth factor inducibility of the pronephric glomus in Xenopus laevis. Development (Cambridge, England) 126:5847–5856
Qiao J, Bush KT, Steer DL, Stuart RO, Sakurai H, Wachsman W, Nigam SK (2001) Multiple fibroblast growth factors support growth of the ureteric bud but have different effects on branching morphogenesis. Mech Dev 109:123–135
Qiao J, Uzzo R, Obara-Ishihara T, Degenstein L, Fuchs E, Herzlinger D (1999) FGF-7 modulates ureteric bud growth and nephron number in the developing kidney. Development (Cambridge, England) 126:547–554
Nguyen HQ, Danilenko DM, Bucay N, DeRose ML, Van GY, Thomason A, Simonet WS (1996) Expression of keratinocyte growth factor in embryonic liver of transgenic mice causes changes in epithelial growth and differentiation resulting in polycystic kidneys and other organ malformations. Oncogene 12:2109–2119
Li Z, Jerebtsova M, Liu XH, Tang P, Ray PE (2006) Novel cystogenic role of basic Fibroblast Growth Factor in developing rodent kidneys. Am J Physiol Renal Physiol 291:F289–296
Ohuchi H, Hori Y, Yamasaki M, Harada H, Sekine K, Kato S, Itoh N (2000) FGF10 acts as a major ligand for FGF receptor 2 IIIb in mouse multi-organ development. Biochem Biophys Res Commun 277:643–649
Sun X, Meyers EN, Lewandoski M, Martin GR (1999) Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes Dev 13:1834–1846
Grieshammer U, Cebrian C, Ilagan R, Meyers E, Herzlinger D, Martin GR (2005) FGF8 is required for cell survival at distinct stages of nephrogenesis and for regulation of gene expression in nascent nephrons. Development (Cambridge, England) 132:3847–3857
Perantoni AO, Timofeeva O, Naillat F, Richman C, Pajni-Underwood S, Wilson C, Vainio S, Dove LF, Lewandoski M (2005) Inactivation of FGF8 in early mesoderm reveals an essential role in kidney development. Development (Cambridge, England) 132:3859–3871
Celli G, LaRochelle WJ, Mackem S, Sharp R, Merlino G (1998) Soluble dominant-negative receptor uncovers essential roles for fibroblast growth factors in multi-organ induction and patterning. EMBO J 17:1642–1655
Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM (1996) Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet 12:390–397
Weinstein M, Xu X, Ohyama K, Deng CX (1998) FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development (Cambridge, England) 125:3615–3623
Revest JM, Spencer-Dene B, Kerr K, De Moerlooze L, Rosewell I, Dickson C (2001) Fibroblast growth factor receptor 2-IIIb acts upstream of Shh and Fgf4 and is required for limb bud maintenance but not for the induction of Fgf8, Fgf10, Msx1, or Bmp4. Dev Biol 231:47–62
Steinberg F, Zhuang L, Beyeler M, Kalin RE, Mullis PE, Brandli AW, Trueb B (2010) The FGFRL1 receptor is shed from cell membranes, binds fibroblast growth factors (FGFs), and antagonizes FGF signaling in Xenopus embryos. J Biol Chem 285:2193–2202
Xu X, Weinstein M, Li C, Naski M, Cohen RI, Ornitz DM, Leder P, Deng C (1998) Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction. Development (Cambridge, England) 125:753–765
Arman E, Haffner-Krausz R, Chen Y, Heath JK, Lonai P (1998) Targeted disruption of fibroblast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development. Proc Nat Acad Sci USA 95:5082–5087
Yamaguchi TP, Harpal K, Henkemeyer M, Rossant J (1994) fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes Dev 8:3032–3044
Deng CX, Wynshaw-Boris A, Shen MM, Daugherty C, Ornitz DM, Leder P (1994) Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes Dev 8:3045–3057
Sims-Lucas S, Argyropoulos C, Kish K, McHugh K, Bertram JF, Quigley R, Bates CM (2009) Three-dimensional imaging reveals ureteric and mesenchymal defects in Fgfr2-mutant kidneys. J Am Soc Nephrol 20:2525–2533
Sims-Lucas S, Cullen-McEwen L, Eswarakumar VP, Hains D, Kish K, Becknell B, Zhang J, Bertram JF, Wang F, Bates CM (2009) Deletion of Frs2alpha from the ureteric epithelium causes renal hypoplasia. Am J Physiol Renal Physiol 297:F1208–1219
Brummer T, Schmitz-Peiffer C, Daly RJ (2010) Docking proteins. FEBS J 277:4356–4369
Eswarakumar VP, Ozcan F, Lew ED, Bae JH, Tome F, Booth CJ, Adams DJ, Lax I, Schlessinger J (2006) Attenuation of signaling pathways stimulated by pathologically activated FGF-receptor 2 mutants prevents craniosynostosis. Proc Nat Acad Sci USA 103:18603–18608
Hains D, Sims-Lucas S, Kish K, Saha M, McHugh K, Bates CM (2008) Role of fibroblast growth factor receptor 2 in kidney mesenchyme. Pediatr Res 64:592–598
Wang Y, Tripathi P, Guo Q, Coussens M, Ma L, Chen F (2009) Cre/lox recombination in the lower urinary tract. Genesis 47:409–413
Hains DS, Sims-Lucas S, Carpenter A, Saha M, Murawski I, Kish K, Gupta I, McHugh K, Bates CM (2010) High incidence of vesicoureteral reflux in mice with Fgfr2 deletion in kidney mesenchyma. J Urol 183:2077–2084
Acknowledgements
The author wishes to thank Elsevier and the American Society of Nephrology for permission to reprint some of the figures used in this publication. Some of the work presented was supported by grants from the National Institute of Diabetes and Digestive and Kidney Disease, R01 DK070030 and R01 DK081128 (C. B.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bates, C.M. Role of fibroblast growth factor receptor signaling in kidney development. Pediatr Nephrol 26, 1373–1379 (2011). https://doi.org/10.1007/s00467-010-1747-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-010-1747-z