
Abstract
Distal renal tubular acidosis (RTA) with nerve deafness is caused by mutations in the ATP6V1B1 gene causing defective function of the H+-ATPase proton pump. We report five acidotic children (four males) from four unrelated families: blood pH 7.21–7.33, serum bicarbonate 10.8–14.7 mEq/l, minimum urinary pH 6.5–7.1 and fractional excretion of bicarbonate in the presence of normal bicarbonatemia 1.1–5.7%. Growth retardation and nephrocalcinosis, but not hypercalciuria, were common presenting manifestations. Hearing was normally preserved in one of the patients whose sister was severely deaf. One child was homozygous for a known mutation in exon 1: C>T (R31X). Three children were homozygous for a splicing mutation, intron 6 + 1G>A. The other patient was a compound heterozygote, having this mutation and a previously unreported mutation in exon 10: G>A (E330K). Our report shows that hearing loss is not always present in the syndrome of distal renal tubular acidosis with nerve deafness and the absence of hypercalciuria at diagnosis and describes a new mutation responsible for the disease in the ATP6V1B1 gene.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary distal renal tubular acidosis is caused by a failure of the α-intercalated cells of the cortical collecting duct to secrete acid into the tubular lumen. Biochemical diagnostic criteria of distal RTA include sustained hyperchloremic metabolic acidosis with an inability to maximally acidify the urine below a pH of 5.5 and the absence of massive bicarbonaturia revealed by normal fractional excretion of bicarbonate in the presence of normal bicarbonatemia. Hypokalemia, hypercalciuria and hypocitraturia are common manifestations of distal RTA. Clinically, the disease is characterized by failure to thrive, nephrocalcinosis, polyuria and urolithiasis. In untreated cases, the progression of nephrocalcinosis may lead to chronic renal failure [1–3].
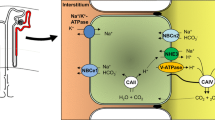
Most cases of primary distal RTA in children result from defective function of the vacuolar H+-ATPase located at the apical surface of the α-intercalated cells. The H+-ATPase is a proton pump formed by several subunits. Mutations in the ATP6V0A4 gene, which encodes the a4 subunit, cause autosomal recessive distal RTA (OMIM #602722) [4]. Mutations in the ATP6V1B1 gene, encoding the B1 subunit, result in autosomal recessive distal RTA associated with nerve deafness (OMIM #267300) [5]. ATP6V1B1 is expressed in the cochlea and the endolymphatic sac, where the H+-ATPase pump likely plays an important physiological role to maintain the endolymph pH at 7.4.
We report five patients with distal RTA having underlying mutations in the ATP6V1B1 gene and discuss the association with hearing loss and the clinical spectrum of this entity.
Patients and methods
Patients
Five Spanish Caucasian children (four males) diagnosed with primary distal RTA according to the clinical and biochemical findings were studied. Hearing was assessed by pure-tone audiometry and/or auditory evoked responses. The data of patient I-1 have been partially reported [5, 6]. The children were members of four non-related families living in the same geographical region of Spain (Asturias). Their parents did not claim consanguinity.
Genetic analysis of ATP6V1B1 gene
DNA was extracted by using the salting-out method [7] from blood samples from the patients and their parents, except for patient IV-1’s father, who was not available. DNA from 100 healthy individuals was used as the control. Informed consent was obtained from all the participants in the study.
The 15 exons of ATP6V1B1 were amplified by polymerase chain reaction (PCR) using the primers shown in Table 1. The amplified fragments were purified and sequenced on an automated ABI310 system, using BigDye chemistry (Applied Biosystems-Applera Corporaton, Drive Foster City, CA).
Results
Clinical and biochemical data
Tables 2 and 3 summarize the clinical and biochemical manifestations leading to the diagnosis of distal RTA in each patient.
ATP6V1B1 analysis
Four children were carriers of a previously reported splicing mutation: intron 6 + 1G>A [5]. Patients II-1, III-1 and III-2 were homozygous for this mutation. Patient IV-1 was a compound heterozygote who had this mutation and a previously unreported mutation in exon 10: G>A (E330K). Genotyping of this mutation by restriction enzyme (Taq I) digestion showed its absence in the 100 controls. Patient I-1 was homozygous for a known mutation in exon 1: C>T (R31X) [5]. The parents were mutation carriers.
Discussion
Our patients had three different ATP6V1B1 gene mutations. The intron 6 + 1 G>A mutation was found homozygously in three patients from two families and in another who was a compound heterozygote. This child also had a mutation E330K, which had not been previously described. Although a functional analysis of the changes induced by this mutation was not performed, replacement of an acidic amino acid, glutamic acid, by a basic one, lysine, likely brings about important conformational alterations in the structure of the protein. Moreover, glutamic acid is an amino acid highly conserved in the composition of this protein among species, which suggests an important biological role. The presence of the splice-site mutation intron 6 + 1 g>a in four of the patients, homozygous in three, from families apparently unrelated might suggest a Spanish founder effect. Table 4 shows ATP6V1B1 gene mutations reported in patients with distal RTA associated with nerve deafness [5, 9–11].
The five patients reported here provide interesting data on the syndrome of distal RTA associated with hearing loss. The two acidotic siblings of family III have the same mutation, but whereas the oldest sister (III-1) was severely deaf, her brother (III-2) hears normally. As this brother was born after his sister had been diagnosed, early and repeated postnatal explorations were performed, revealing metabolic acidosis in the 1st weeks of life and unimpaired hearing. Subsequently, he acquired language at a normal age. To our knowledge, systematic newborn hearing screening in patients later diagnosed with distal RTA with nerve deafness has not been reported, and the hearing status in the early postnatal period is unknown for these patients. We have previously reported patient I-1 and his dizygotic twin, who was deaf, but not acidotic [6]. The patients from these two families illustrate the clinical heterogeneity associated with ATP6V1B1 mutations, ranging from severe deafness to normal audition, at least in the early stages. Patient III-2 had been treated since the 1st weeks of life with well-controlled bicarbonate supplementation. Sustained correction of the acidosis may have had a protective effect on the development and progression of deafness in this patient. Although it has been suggested that alkali treatment does not prevent the progression of hearing impairment [12], the late development of hearing loss in some patients with distal RTA linked to ATP6V0A4 mutations might be related with inappropriate sustained control of acidosis.
Metabolic acidosis with reduced excretion of fixed acids and positive hydrogen ion balance leads to hypercalciuria. It has been proposed that in patients with primary distal RTA there is a significantly negative correlation between the serum bicarbonate concentration and urine calcium elimination, whereby normal values of urinary excretion of calcium may be used as a marker of well-controlled acidosis [13]. In this respect, it is interesting to note that our patients were normocalciuric at the time of diagnosis, in spite of the presence of simultaneous metabolic acidosis (Table 3). Levels of urinary calcium elimination are normally high in infants and decrease progressively as the child becomes older. Therefore, age must be strictly considered at the time of evaluating calcium excretion in the urine. In addition, the development of nephrocalcinosis and urolithiasis in the absence of hypercalciuria supports the important pathogenic role of hypocitraturia in the origin of these complications.
After a follow-up from 5 to 29 years, the five patients reported here had maintained normal glomerular filtration rates, persistent nephrocalcinosis, the need for a hearing prosthesis in the four cases with hearing impairment and growth improvement following alkali supplementation. This confirms the good prognosis of distal RTA when metabolic acidosis has been well controlled since an early age.
References
Roth KS, Chan JC (2001) Renal tubular acidosis: a new look at an old problem. Clin Pediatr (Phila) 40:533–543
Caldas A, Broyer M, Dechaux M, Kleinknecht C (1992) Primary distal tubular acidosis in childhood: clinical study and long-term follow-up of 28 patients. J Pediatr 121:233–241
Laing CM, Toye AM, Capasso G, Unwin RJ (2005) Renal tubular acidosis: developments in our understanding of the molecular basis. Int J Biochem Cell Biol 37:1151–1161
Karet FE (2002) Inherited distal renal tubular acidosis. J Am Soc Nephrol 13:2178–2184
Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez-Soriano J, Santos F, Cremers CW, Di Pietro A, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skvorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP (1999) Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 21:84–90
Santos F, Rey C, Malaga S, Rodriguez LM, Orejas G (1991) The syndrome of renal tubular acidosis and nerve deafness. Discordant manifestations in dizygotic twin brothers. Pediatr Nephrol 5:235–237
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215
Matos V, van Melle G, Boulat O, Markert M, Bachmann C, Guignard JP (1997) Urinary phosphate/creatinine, calcium/creatinine, and magnesium/creatinine ratios in healthy pediatric population. J Pediatr 131:252–257
Stover EH, Borthwick KJ, Bavalia C, Eady N, Fritz DM, Rungroj N, Giersch AB, Morton CC, Axon PR, Akil I, Al-Sabban EA, Baguley DM, Bianca S, Bakkaloglu A, Bircan Z, Chauveau D, Clermont MJ, Guala A, Hulton SA, Kroes H, Li Volti G, Mir S, Mocan H, Nayir A, Ozen S, Rodriguez Soriano J, Sanjad SA, Tasic V, Taylor CM, Topaloglu R, Smith AN, Karet FE (2002) Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet 39:796–803
Hahn H, Kang HG, Ha IS, Cheong HI, Choi Y (2003) ATP6B1 gene mutations associated with distal renal tubular acidosis and deafness in a child. Am J Kidney Dis 41:238–243
Vargas-Poussou R, Houillier P, Le Pottier N, Strompf L, Loirat C, Baudouin V, Macher MA, Dechaux M, Ulinski T, Nobili F, Eckart P, Novo R, Cailliez M, Salomon R, Nivet H, Cochat P, Tack I, Fargeot A, Bouissou F, Kesler GR, Lorotte S, Godefroid N, Layet V, Morin G, Jeunemaitre X, Blanchard A (2006) Genetic investigation of autosomal recessive distal renal tubular acidosis: evidence for early sensorineural hearing loss associated with mutations in the ATP6V0A4 gene. J Am Soc Nephrol 17:1437–1443
Zakzouk SM, Sobki SH, Mansour F, al Anazy FH (1995) Hearing impairment in association with distal renal tubular acidosis among Saudi children. J Laryngol Otol 109:930–934
Rodriguez-Soriano J, Vallo A, Castillo G, Oliveros R (1982) Natural history of primary distal renal tubular acidosis treated since infancy. J Pediatr 101:669–676
Ackowledgements
This study was supported by Red de Centros de Genética Clínica y Molecular (C03-07) del Instituto de Salud Carlos III, FIS PI 051261, Fondos FEDER and Fundación Nutrición y Crecimiento.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gil, H., Santos, F., García, E. et al. Distal RTA with nerve deafness: clinical spectrum and mutational analysis in five children. Pediatr Nephrol 22, 825–828 (2007). https://doi.org/10.1007/s00467-006-0417-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-006-0417-7