
Abstract
Renal artery stenosis (RAS) is one of the most common causes of severe arterial hypertension in infants. Its management is very difficult, especially when present in a single kidney. We report a case of severe hypertension caused by RAS of congenital single pelvic kidney in a 4-month-old boy. The patient presented with cardiorespiratory insufficiency that was first treated as acute fulminate myocarditis. Medical treatment of arterial hypertension was disappointing, as it had to be balanced between congestive cardiac failure and acute renal failure. Percutaneous transluminal angioplasty (PTA) done by coronary balloon dilatation catheters through the left axillary access was successful. Following dilatation of the renal artery, blood pressure decreased and its good control was possible by only one drug. With improved medical blood pressure control and normal growth development, the reassessment of clinical therapy options adjusted to a larger vessel size would be possible. Renovascular hypertension due to RAS in infants with a solitary kidney is difficult to control by medical treatment alone. PTA should be considered as a viable option in infants with refractory hypertension due to renal artery stenosis in a solitary kidney, since it has the potential of improving hypertension while preserving renal function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Arterial hypertension is rare in infancy [1] and usually secondary to an underlying disease. The most common of these diseases are coarctation of the aorta, renal artery stenosis and renal parenchymal disease [2]. In most cases, hypertension in infants presents with nonspecific signs—such as irritability, lethargy, failure to thrive, and respiratory distress—that are related to the complications of severe hypertension, including congestive heart failure. Some patients may be dehydrated due to hyponatremic hypertensive syndrome [3]. The potential morbidity and mortality of untreated hypertension in infancy is high. At this age group, antihypertensive management poses a difficult challenge due to the small size of infant’s renal vessels limiting the options for complete cure by percutaneous transluminal angioplasty (PTA) or reconstructive vascular surgery [4]. Infants with bilateral renal artery stenosis or those with renal artery stenosis of a solitary functioning kidney are the most vulnerable groups. In these patients, antihypertensive agents are either ineffective in controlling severe hypertension or have the potential for acute renal ischemia and other adverse effects [5]. In such children a minimally invasive treatment to preserve glomerular filtration rate and to limit vital organ injury due to systemic hypertension is unquestionably attractive.
Here we report an interesting case of an infant with severe renal-artery stenosis of a single congenital pelvic kidney and discuss the methodological approach to the diagnosis and antihypertensive treatment in such patients.
Case report
A 4-month-old male was referred to the emergency department because of severe respiratory distress. He had been admitted to a local hospital 3 days prior with a 1-day history of dyspnoea and cough without fever. In the local hospital he was unsuccessfully treated as having acute bronchiolitis with intravenous aminophylline, intravenous corticosteroids and penicillin. Blood pressure (BP) was not measured. As the child’s condition rapidly deteriorated, he was started on mechanical ventilation and transferred to the University Children’s Hospital, where he was admitted to the pediatric intensive care unit (PICU).
The child had an unremarkable prenatal and family history. He was born by vaginal delivery by foot presentation. Birth weight was 2,700 g and Apgar score was 8 at 1 min and 5 min. In the early postnatal period central nervous ultrasound examination showed a mild degree of periventricular and intraventricular hemorrhages. Otherwise, the child was considered healthy.
Hospital course
Upon physical examination on admission to the PICU, the child looked acutely sick, pale and edematous, and he was dependent upon mechanical ventilatory support. He was found to have a lot of pulmonary crackles, tachycardia and a mild systolic ejection murmur along the left upper sternal border. His weight was 5.5 kg (fifth percentile). Initially, BP was 130/98 mmHg, but subsequently during the next 3 h it decreased, with systolic BP ranging between 94 mmHg and 100 mmHg and diastolic BP between 67 mmHg and 77 mmHg. However, during further follow-up, BP gradually increased to an average of 160/100 mmHg and maximal values of 220/160 mmHg.
Initial investigation revealed normal blood count, normal serum electrolytes but a slightly decreased serum sodium (131 mmol/l), mildly increased serum urea (7.7 mmol/l) and creatinine (56 μmol/l). The serum lactate dehydrogenase was increased (1,053 U/l, normal less than 460) as was creatinine kinase CK-MB mass (14.6 μg/l, normal less than 0.5 μg/l), troponin (0.6 μg/l, normal less than 0.1 μg/l) and C-reactive protein (34 mg/l, normal less than 5 mg/l). Urinalysis showed 1+ protein by sulfosalicylic acid, while the ratio of protein and creatinine was 0.27 mg/mg. All bacterial and viral cultures were negative. The patient’s urinary output ranged from 580–710 ml per day. A chest X-ray showed cardiac chamber enlargement and the presence of pulmonary edema. Echocardiogram documented severe cardiac dilatation, biventricular hypertrophy and moderate insufficiency of mitral and aortal valves.
Based on the above findings, the patient was diagnosed as having fulminant myocarditis and was treated with: intravenous gamma globulin 1 g/kg during 5 days, milrinone one infusion of 300 µg, methylprednisolone 30 mg/kg during 6 days, captopril 0.1–0.2 mg/kg t.i.d., furosemide 1 mg/kg 3 t.i.d., nifedipine 0.2 mg/kg t.i.d., ranitidine 30 mg daily and meronem 20 mg/kg t.i.d. during 10 days.
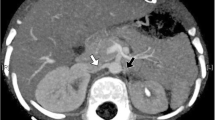
After 3 days the child’s condition improved; mechanical ventilation was discontinued on the fourth day, and the patient was transferred to the cardiology department. At this point, he was thought to be hypertensive secondary to the treatment with intravenous corticosteroids. However, as the hypertension (mean BP 150/89 mmHg, maximal BP 170/120 mmHg), resistant to multiple antihypertensive therapies consisting of captopril, nifedipine and labetalol at maximal doses, persisted after interrupted corticosteroids, steroid-induced hypertension was questioned and the patient was transferred to the nephrology department for further investigation and treatment. Abdominal ultrasound revealed a solitary kidney (63×28 mm) lying just behind the urinary bladder, which was confirmed by intravenous urography. Renal Doppler sonography showed no abnormalities. On voiding cystography, vesicoureteral reflux grade II was discovered. Renal scintigraphy revealed no scarring. Abdominal aortography disclosed a solitary, ectopic kidney with severe stenosis of renal artery (diameter about 0.8 mm) branching from the aortic bifurcation at a sharp angle (Fig. 1a). At this point, due to a very high risk of acute renal failure, which can develop in patients with renal artery stenosis, captopril therapy was discontinued and prazosin (0.06 mg/kg b.i.d.) was added to the antihypertensive regimen. The patient continued to be severely hypertensive (mean BP 157/89 mmHg, maximal BP 220/110 mmHg), and cardiac failure manifested again as a severe respiratory distress. Captopril was reintroduced with diuretic, but serum creatinine increased to 58 μmol/l. The decision was made to attempt percutaneous transluminal angioplasty with surgical stand-by and anesthesiologic support. The procedure was performed by a vascular interventional radiologist via the left axillary artery approach. The stenotic segment of renal artery was dilated using progressively larger balloons designed for the dilatation of coronary arteries (3.0×20 mm and later 4.0×20 mm Maveric Monorail Balloon catheter, Boston Scientific). Control abdominal aortography demonstrated increased renal artery diameter of about 400% (Fig. 1b) and better opacification of the kidney. For anticoagulation, the child received 50 IU heparin/kg intravenously at the beginning of the procedure, and after removal of the sheath 10 IU heparin/kg per hour continuously for 48 h, followed by Fraxiparin 70 IU/kg per day during the next 10 days for thrombosis prophylaxis. Following dilatation of the renal artery, blood pressure immediately decreased to 110/70 mmHg and all antihypertensive drugs were stopped. However, to achieve sustained blood pressure values under 120/80 mmHg, medical therapy (labetalol 7 mg/kg/day) was needed. As blood pressure was brought under control, the signs and symptoms of congestive heart failure resolved. Doppler sonography revealed no evidence of recurrent renal-artery stenosis. Laboratory examination showed normal electrolytes and normal serum levels of urea and creatinine (34 μmol/l). The patient was discharged from hospital 10 days after the procedure with blood pressure 110–120/70–80 mmHg and is currently on oral labetalol and antibacterial prophylaxis due to vesicoureteral reflux. He was seen in the outpatient clinic every 2 weeks. During the next 3 months, he gained 2.5 kg in body weight, and his height increased 7 cm.

a Abdominal aortography shows severe stenosis of renal artery (diameter about 0.8 mm) (white arrow) branching from the aortic bifurcation at a sharp angle; b Successful angioplasty of renal artery stenosis (white arrow)
Discussion
This case report is instructive from several points of view. The first is unrecognized severe hypertension in an infant whose case was presumptively diagnosed as severe bronchiolitis in a local hospital and as fulminate myocarditis in the PICU of the University Children’s Hospital. Being uncommon in infants, arterial hypertension is rarely diagnosed until it becomes symptomatic. Congestive heart failure is a common manifestation of severe hypertension in infants [2,4,5,6,7,8]. Its early signs, such as irritability, feeding problems and mild respiratory distress, can progress very quickly to severe cardiac insufficiency. Therefore, in such infants one must consider severe arterial hypertension in the differential diagnosis of congestive heart failure, and one must measure blood pressure. Normal blood pressure or blood pressure in the upper normal range after resuscitation must alert the clinician to the possibility of severe hypertension as responsible for congestive heart failure [8]. A rational approach to reveal the cause of severe hypertension in infants includes renal ultrasound as initial screening for renal parenchymal and urological abnormalities, color and duplex Doppler sonography for rapid and noninvasive screening of renal vessel abnormalities, and renal isotope studies before and after angiotensin-converting enzyme inhibitors (ACEI) to detect a hemodynamically significant renovascular disease [9]. While some cases of renovascular lesions in infants may be diagnosed by Doppler ultrasound, other cases, including ours, have demonstrated that this technique may easily miss the diagnosis of renovascular hypertension [9,10]. Therefore, a negative or “normal” Doppler examination should be followed by renal isotope scans and/or angiography. Also, severe hypertension or failure to control hypertension effectively with one drug is a golden indication that should guide the physician to apply renal arteriography to detect renovascular disease [11]. CT angiography and MR angiography, although promising in the adult population [12], still have limited application in infants [9].
Our case gave us valuable information regarding the vascularization of the congenital pelvic kidney and management of renovascular hypertension in infants with solitary kidney. The congenital pelvic kidney results from a failure of renal ascents during the fourth to eighth week of gestation [13]. The blood supply in the pelvic kidney presents with more anatomic variation than in the normally positioned lumbar kidney [13,14,15,16,17]. The most common abnormal vascular supply consists of one or two renal arteries that arise from the distal aorta, the bifurcation, and the common or external iliac artery [16]. In the case reported here, the right renal artery arose from the bifurcation of aorta. To the best of our knowledge, RAS and single pelvic kidney has been rarely reported [18]. This is potentially a harmful condition associated with severe hypertension, progressive renal failure, and a high risk of morbidity and mortality. In infants, the control of BP with antihypertensive drugs is the method of choice, to enable normal growth development until surgical correction of the renal artery stenosis has a chance to be successful [4]. However, the refractoriness to medical therapy, as was found in our patient, is common. Its pathogenesis can be explained by the model of one-kidney, one-clip Goldblatt hypertension, in which renal hypoperfusion leads to hyper-activation of the renin-angiotensin-aldosterone system with a consequent circulatory volume expansion that cannot be modulated by pressure natriuresis by means of the unaffected kidney. Pharmacological blockade of the renin-angiotensin-aldosterone system may result in acute renal failure [4]. Therefore, medical therapy accompanied by little compensatory renin release and minimal effect on glomerular filtration rate, such as labetalol and alpha-adrenergic blocking agents, is preferred. PTA is less invasive than open surgery, but the overall experience in small children is limited [5,19]. Potential complications of PTA include hemorrhage or hematoma at the puncture site, transient renal failure induced by contrast media, spasm of the renal artery, embolization of the kidney, renal infarction, dissection, perforation or rupture of the renal vessels, anuria and balloon rupture. Our experience is encouraging, as PTA using low profile coronary artery balloon dilatation catheters and the most suitable vascular approach could be performed even in very young infants. Although our patient is currently gaining weight and developing normally, these parameters along with his BP will need to be monitored frequently until his physical size is adequate for eventual vascular repair.
Conclusion
Renovascular hypertension due to RAS in infants with a solitary kidney is difficult to control by medical treatment alone. PTA should be considered as a viable option in infants with refractory hypertension due to renal artery stenosis in a solitary kidney.
References
Goble MM (1993) Hypertension in infancy. Pediatr Clin North Am 40(1):105–122
Ingelfinger JR (2002) Hypertension. In: Edelmann CM Jr (ed) Pediatric kidney disease, 2nd edn. Little Brown, Boston, pp 1889–1908
Peco-Antic A (2005) Pediatric hypertensive syndrome. Therapy 2:301–309
Bendel-Stenzel M, Najarian JS, Sinaiko AR (1996) Renal artery stenosis in infants: long-term medical treatment before surgery. Pediatr Nephrol 10:147–151
Ellis D, Shapiro R, Scantlebury VP, Simmons R, Towbin R (1995) Evaluation and management of bilateral renal artery stenosis in children: a case series and review. Pediatr Nephrol 9:259–267
Dixon BP, Devarajan P, Mitsnefes M (2005) Neonatal renovascular hypertension due to prenatal traumatic retroperitoneal hematoma. Pediatr Nephrol 20:670–672
Takigiku K, Satomi G, Yasukochi S (2002) Successful transluminal angioplasty of renal arterial stenosis using the transcarotid approach. Cardiol Young 12:589–591
Kovacikova L, Kunovsky P, Skrak P, Haviar D, Martanovic P (2005) Renovascular hypertension in infant presenting with cardiogenic shock. Pediatr Emerg Care 21(5):322–324
Roth CG, Spottswood SE, Chan JC, Roth KS (2003) Evaluation of the hypertensive infant: a rational approach to diagnosis. Radiol Clin North Am 41:931–944
Cochat F, Bogaru A, MIcheli JL, Lepori D, Guignard JP (2004) Severe hypertension and massive proteinuria in a newborn with renal artery stenosis. Pediatr Nephrol 19:544–546
Shahdadpuri J, Frank R, Gauthier BG, Siegel DN, Trachtman H (2000) Yield of renal arteriography in the evolution of pediatric hypertension. Pediatr Nephrol 14:816–819
Schoenberg SO, Rieger J, Johannson LO, Dietrich O, Bock M, Prince MR, Reiser MF (2003) Diagnosis of renal artery stenosis with magnetic resonance angiography: update 2003. Nephrol Dial Transplant 18:1252–1256
Hollis HW, Rutherford RB, Crawford GJ, Cleland BP (1989) Abdominal aortic aneurysm repair in patients with pelvic kidney. Technical considerations and literature review. J Vasc Surg 9:404–409
Sebe P, Chemla E, Varkarakis J, Latremouill C (2004) Anatomic variations of the vascularization of the pelvic kidney: apropos of a case and review of the literature. Morphologie 88:24–26
Gulsun M, Balkanci F, Cekirge S, Deger A (2000) Pelvic kidney with an unusual blood supply: angiographic findings. Surg Radiol Anat 22:59–61
Glock Y, Blasevich R, Laghzaoui A, Roux D, Fournial G (1997) Abdominal aortic aneurysm and congenital pelvic kidney. A rare association. Tex Heart Inst J 24(2):131–133
Murakami T, Makino Y, Suto Y, Yasuda K (2004) Abdominal aortic aneurismal repair in a patient with a congenital solitary pelvic kidney. A case report. J Cardiovasc Surg 45:501–504
Dean P, Brancaccio C, Recchia G, Prati GF, Gulino S, Ancona G (1984) Case of renovascular hypertension caused by renal artery stenosis in congenital single kidney in pelvic ectopy. Minerva Urol Nefrol 36:135–138
Hofbeck M, Singer H, Rupprecht T, Ruder H, Schmiedl N (1998) Successful percutaneous transluminal angioplasty for treatment of renovascular hypertension in a 15-month-old child. Eur J Pediatr 157:512–514
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Peco-Antic, A., Djukic, M., Sagic, D. et al. Severe renovascular hypertension in an infant with congenital solitary pelvic kidney. Pediatr Nephrol 21, 437–440 (2006). https://doi.org/10.1007/s00467-005-2109-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-005-2109-0