
Abstract
Immune complex and complement systems play an important role in membranoproliferative glomerulonephritis (MPGN). X-linked agammaglobulinemia (XLA) is a primary immunodeficiency characterized by severe hypogammaglobulinemia. We report the case of an XLA patient who developed MPGN during an intravenous immunoglobulin (IVIG) treatment. In this patient, the serum IgG level was maintained at more than 400 mg/dl of regular IVIG administration (2.5 g/dose/month). The patient presented with microscopic hematuria, proteinuria (U-pro/Cr: 4.0–4.2) and low serum complement levels (C3: 57.8 mg/dl) 3 years after IVIG treatment and was diagnosed histopathologically as having MPGN type III. Both hematuria and proteinuria significantly improved, and the serum complement level returned to a normal level following methylprednisolone pulse therapy. To our knowledge, this is the first case report of MPGN associated with XLA. Although it is unclear how MPGN occurred in this XLA patient, we suggest that residual humoral immunity in the patient could be associated with the development of MPGN.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In some cases of membranoproliferative glomerulonephritis (MPGN), immune complex and complement systems can play important roles. The glomerular deposition of immune complexes and the activation of complement systems cause glomerular inflammation, which may result in MPGN [1, 2, 3, 4]. X-linked agammaglobulinemia (XLA) is a primary immunodeficiency characterized by an extreme reduction in the number of B cells, resulting in severe hypogammaglobulinemia. A mutation in the Bruton’s tyrosine kinase ( BTK) gene on the X chromosome has been known to be responsible for this disease [5, 6]. Most children with XLA contract severe bacterial infections during late infancy, and the disease is of a life-threatening nature that requires intravenous immunoglobulin (IVIG) replacement therapy to maintain serum levels of IgG at greater than 400 mg/dl. We report a case of MPGN that developed during the course of IVIG replacement therapy in a patient with XLA.
Case report
The patient in this report was a 2-month-old boy. The patient’s older brother was admitted to our hospital because of bacterial pneumonia when he was 20 months old. The older brother’s serum IgG level was found to be at an undetectable level, and the number of peripheral B cells was diminished (1%). Despite the absence of a family history of immunodeficiencies, it was suggested that he had XLA. A genetic analysis was performed and disclosed a mutation (612insA) in the BTK gene. Genetic analyses of the BTK gene in his family members showed the same mutation in his mother and the patient (aged 2 months), indicating that they were a carrier of XLA and had asymptomatic XLA, respectively. The patient showed detectable levels of serum IgG, IgA and IgM since early infancy (152–242 mg/dl: normal range, 311–449 mg/dl, IgA 12.1–37.2 mg/dl: normal, 34-120 mg/dl, IgM 4.2–30.1 mg/dl: normal, 42–74 mg/dl). Meanwhile, the older brother did not show a detectable level of serum immunoglobulin. IVIG replacement therapy was initiated at the age of 20 months in the elder brother (200 mg/kg/month) and at the age of 2 months in the patient (300 mg/kg/month). IVIG replacement therapy effectively maintained serum IgG levels above 400 mg/dl in both brothers.
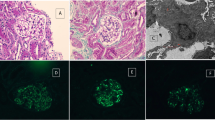
At the age of 3 years, the patient presented with microscopic hematuria, mild proteinuria (U-pro/Cr: 0.83) and low serum complement (C3: 58.7 mg/dl: normal range 65–135 mg/dl). The amounts of IVIG required to maintain adequate serum levels of IgG gradually increased (up to 7.5 g/month). His proteinuria was increased (U-pro/Cr: 4.0–4.2) and low serum complement continued (C3:57.8 mg/dl) (Fig. 1). Renal biopsy was performed at the age of 4 years. Histopathology showed that the obtained glomeruli were enlarged and lobulated. Diffuse and global proliferation of mesangial cells, mildly increased mesangial matrix, thickening of the glomerular basement membrane (GBM) and mesangial interposition were noted. There were no interstitial changes. Immunofluorescence (IF) microscopy revealed strong staining of IgG, C3c and C3d in granular form within the mesangium and GBM and a weak staining of C1q, C4 and IgA within the mesangium and GBM, but IgM was negative. In electron microscopic observation, effacement of the foot process and abundant subendothelial deposits together with some paramesangial and subepithelial deposits were also observed (Fig. 2). The patient was diagnosed as having MPGN type III. His renal function remained normal (his serum creatinine ranged from 0.2 to 0.4, and his creatinine clearance ranged from 150 to 250 ml/min/1.73 m2). He had normal liver function and negative serum hepatitis B virus surface antigen and hepatitis C virus (HCV) antibody. The elder brother with XLA had no hematuria or proteinuria and had a normal level of serum complement (100–140 mg/dl).

Clinical course of the patient. *Dose of IVIG, **methylprednisolone pulse therapy, ***prednisolone, †serum IgG levels before administration of IVIG on the respective days, ‡normal range at this patient’s age

Electron microscopic findings of the kidney of the patient. Effacement of foot process was observed, subendothelial deposits were prominent, and some paramesangial and subepithelial deposits were seen. E Endothelial cell, Ep epithelial cell, M mesangial cell, Δ GBM glomerular basement membrane, ⇧ subendothelial deposits, heavy filled arrow paramesangial deposits, ⌂ subepithelial deposits, ↑ effacement of foot process, ⇑ mesangial deposits
Despite the use of three different preparations (Venilon®, Venoglobulin-IH® and Gammaguard®), hematuria and proteinuria did not improve. The serum level of IgG decreased further, which required an increase in IVIG dose. Immune complex was measured by three different methods (C1q, monoclonal RF and anti-C3d antibody). Although C1q and monoclonal RF were not detected, anti-C3d antibody was 10.1 µg/ml (normal range, <13 µg/ml). There was no cryoglobulinemia.
Methylprednisolone pulse therapy (MPT) was performed at a dose of 30 mg/kg/day for 3 consecutive days at the age of 5 years, followed by oral prednisolone therapy at a dose of 1 mg/kg/dose every other day. Hematuria and proteinuria improved following MPT therapy. The serum complement levels were normalized, and the serum level of IgG was maintained at a level greater than 400 mg/dl. As a result, the IVIG replacement dose was reduced (Fig. 1). The anti-C3d antibody became undetectable.
Discussion
Since XLA patients produce extremely low levels of immunoglobulin, these patients seem to be less susceptible to chronic immune complex and complement-mediated nephritis such as MPGN. This patient with XLA developed MPGN during IVIG treatment. To the best of our knowledge, this is the first case report of nephritis in an XLA patient.
HCV might be one of the major causes of idiopathic MPGN, and the possibility of HCV infection should be considered. However, for this patient, the liver function test was normal, and HCV antibody was negative, indicating that he did not have HCV-associated MPGN.
Histopathologically, IgG was stained moderately at the glomeruli under IF, while it was uncertain whether the IgG was exogenous (IVIG) or endogenous. There are various possibilities for the pathogenesis of MPGN. Firstly, this deposit might be formed through the reaction of IVIG with any endogenous antigen. Secondly, this deposit might be formed thorough the reaction with the patient’s own endogenous IgG. Unfortunately, we were unable to estimate precisely his innate production of IgG because IVIG treatment was started before the disappearance of maternal IgG at the age of 2 months. Small amounts of IgA and IgM were detected in his serum, which do not cross the placenta, suggesting that the patient has a residual ability to produce his own immunoglobulin. It is conceivable that he can produce IgG as well as IgA and IgM, because it is known that a small number of B cells are present in the peripheral blood of some patients with XLA. These B cells can proliferate, undergo isotype switching and differentiate into specific antibody-producing cells [7]. Wegmüller [8] reported that IVIG might moderately activate a complement system in patients with primary humoral immunodeficiencies as well as in normal individuals. This activation of the complement system might cause MPGN.
We attempted to treat the MPGN using different preparations of IVIG, since MPT might adversely affect an immunocompromised patient. At first, if he produced antibody against IVIG, his disease could improve by using the Fab type of IVIG, which is not an intact type. However, we abandoned this method because of the shortness of its half-life and lower efficacy. Secondly, some substances that are combined with IVIG during its preparation, or even a trace of IgA contained within IVIG, could cause nephritis. Therefore, we chose to use several different types of immunoglobulin preparations. Employment of other preparations (Venilon® treated by sulfonation; Venoglobulin-IH® treated by polyethylene glycol; Gammaguard® treated with ion exchange resins) did not improve the hematuria and proteinuria.
We could not clarify the pathogenesis of MPGN in the patient. Although he and his brother had the same mutation in the BTK gene, only the patient developed MPGN. In this patient, MPT was effective for the treatment of MPGN. These results suggest that a residual humoral immunity in the patient might be causally related to the development of his MPGN.
References
Johnson RJ, Alpers CE, Schen P (2000) Membranoproliferative glomerulonephritis and cryoglobulinemic glomerulonephritis. In: Johnson RJ, Feehally J (eds) Comprehensive clinical nephrology, 1st edn. Mosby Company, St Louis, pp1–10
Holley KE, Donadio JV (1994) Membranoproliferative glomerulonephritis. In: Tisher CC, Brenner BM (eds). Renal pathology, with clinical and functional correlations, vol.1, 2nd edn. JB Lippincott, Philadelphia, pp 284–329
Nakopoulou L (2001) Membranoproliferative glomerulonephritis. Nephrol Dial Transplant 16:71–73
Meyers KEC, Finn L, Kaplan BS (1998) Membranoproliferative glomerulonephritis type III. Pediatr Nephrol 12:512–522
Vetrie D, Vorechovsky I, Sideras P, Holland J, Davies A, Flinter F, Hammarstrom L, Kinnon C, Levinsky R, Bobrow M, et al (1993) The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature 361:226–233
Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, Sparkes RS, Kubagawa H, Mohandas T, Quan S, et al (1993) Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 72:279–290
Nonoyama S, Tsukada S, Yamadori T, Miyawaki T, Jin YZ, Watanabe C, Morio T, Yata J, Ochs HD (1998) Functional analysis of peripheral blood B cells in patients with X-linked agammaglobulinemia. J Immunol 161:3925–3929
Wegmüller E (1998) Effect of intravenous immunoglobulin therapy on plasma complement. Transfus Sci 19:307–318
Acknowledgements
This study was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and the Ministry of Health, Labor and Welfare of Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yoshino, A., Honda, M., Kanegane, H. et al. Membranoproliferative glomerulonephritis in a patient with X-linked agammaglobulinemia. Pediatr Nephrol 21, 36–38 (2006). https://doi.org/10.1007/s00467-005-2029-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-005-2029-z