
Abstract
Chronic hypokalemia is known to induce renal cyst formation in some diseases including primary aldosteronism, distal renal tubular acidosis, Liddle disease and apparent mineralocorticoid excess syndrome. Although chronic hypokalemia is the main clinical feature of Bartter syndrome, renal cyst formation in this disease has never been reported. We describe a patient with classic Bartter syndrome who exhibited renal cysts and nephrocalcinosis. Direct sequencing analysis of the chloride channel CLC-Kb gene identified a heterozygous nonsense mutation (W610X) in exon 16 indicating a diagnosis of Bartter syndrome type III. Although the precise mechanism underlying the development of renal cysts in our patient remains unclear, chronic hypokalemia and nephrocalcinosis may contribute to cyst development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bartter syndrome is a heterogeneous disorder characterized by renal salt wasting, hypokalemic metabolic alkalosis and elevated renin and aldosterone levels with normal or low blood pressure [1]. Recently, five genes have been identified as causing Bartter and Bartter-like syndromes, including genes for a renal bumetanide-sensitive Na-K-2Cl co-transporter, an ATP-sensitive inwardly rectifying K channel (ROMK), a renal chloride channel (ClC-Kb), a β-subunit of ClC-K (barttin) and a calcium-sensing receptor (CASR) [1]. Among them, loss-of-function mutations in the genes of the Na-K-2Cl co-transporter, ROMK and ClC-Kb (CLCNKB) are classified as Bartter syndrome types I, II and III, respectively [1].
Although some phenotypic differences are found in Bartter and Bartter-like syndromes, hypokalemia is the predominant finding of these disorders [2]. Chronic hypokalemia can induce renal structural and functional abnormalities including a concentrating defect, interstitial fibrosis and/or chronic inflammation, and renal cyst formation [3, 4]. Renal cyst formation as a consequence of chromic hypokalemia has been reported in primary aldosteronism [3], primary renal potassium wasting [3], primary distal renal tubular acidosis [5], Liddle syndrome [6] and apparent mineralocorticoid excess syndrome [6, 7]. However, renal cyst formation in Bartter syndrome has never been reported. We describe a patient with Bartter syndrome type III who exhibited renal cysts and nephrocalcinosis.
Case report
A 6-year-old girl was referred to our hospital because of polyuria and polydipsia. The patient was born at 40 weeks of gestation, and her birth weight was 3.1 kg. The prenatal course was unremarkable with no polyhydramnios. The family history was unremarkable. The patient had suffered from polyuria and polydipsia since 2 years of age.
Physical examination on admission revealed a short stature (104.7 cm, −2.3 SD) with appropriate proportion. Her body weight was 15 kg (−1.7 SD) and blood pressure was 110/55 mmHg.
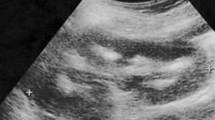
Laboratory studies revealed metabolic alkalosis (pH 7.459 and HCO3 32.1 mmol/l), hypokalemia (2.3 mEq/l) in the presence of an inappropriately high potassium excretion [fractional excretion of potassium (FEK) 20%], hypochloremia (96 mEq/l), an elevated plasma renin activity (16.9 ng/ml/h, normal 0.3–2.9 ng/ml/h) and hypercalciuria (urinary calcium/creatinine ratio 1.09, normal <0.25). Serum concentrations of sodium (140 mEq/l), calcium (9.3 mg/dl), magnesium (2.5 mg/dl, normal 1.8–2.6), phosphate (5.1 mg/dl) and creatinine (0.3 mg/dl), blood urea nitrogen (9.9 mg/dl) level and plasma aldosterone level (5.0 ng/dl, normal 3.6–24.0 ng/dl) were normal. Urinalysis showed no abnormalities. The urinary chloride level was high (105 mEq/l) despite the hypochloremia. Direct sequencing analysis of the CLCNKB using previously described methods [8] identified a heterozygous nonsense mutation (W610X) in exon 16. Renal computed tomography (CT) and renal ultrasound revealed bilateral renal cysts and nephrocalcinosis (Fig. 1).

Post-contrasted renal computed tomography showing bilateral renal cysts. B Bilateral nephrocalcinosis and a small cyst of the left kidney are found
The diagnosis of classic and type III Bartter syndrome with renal cysts and nephrocalcinosis was made. The patient was treated with potassium supplementation, indomethacin and spironolactone, and this treatment partially corrected her hypokalemia (3.2–3.6 mEq/l) and metabolic alkalosis. Four years later, repeated renal CT scan remained an unchanged appearance with renal cysts and nephrocalcinosis.
Discussion
Although Bartter syndrome is an autosomal-recessive disorder, our patient with classic Bartter syndrome revealed only a heterozygous mutation of CLCNKB. Simon et al. [8] and Konrad et al. [9] also described a heterozygous mutation of CLCNKB in some patients with Bartter syndrome, and this might be due to the limitation of gene analyzing methodology.
Our patient exhibited bilateral renal cysts. Renal cyst formation has been reported in some diseases exhibiting chromic hypokalemia. Torres et al. demonstrated that 44% of patients with primary aldosteronism had renal cysts [3]. Igarashi et al. reported that 70.6% of patients with primary distal renal tubular acidosis manifested renal cyst formation [5, 10]. Other diseases with chronic hypokalemia that develop renal cysts include Liddle syndrome [6], apparent mineralocorticoid excess syndrome [6, 7] and primary renal potassium wasting [3]. Although chronic hypokalemia is the predominant feature of Bartter syndrome, this patient had the first reported case of renal cyst formation in this disease.
Although the precise pathogenic mechanisms underlying renal cyst formation in chronic hypokalemia remain unclear, two potential mechanisms have been postulated, including increased renal cell growth and interstitial injury caused by increased production of ammonia [3, 4].
In a rat model, chronic hypokalemia induced hypertrophy and hyperplasia of the epithelial cells in the collecting duct and tubular dilatation because of luminal obstruction by hyperplastic epithelial cells [4]. In addition, an experimental study demonstrated that a decreased extracellular potassium concentration increased the growth of kidney cells in vitro [11]. Although renal tubular epithelial cell hypertrophy or hyperplasia due to hypokalemia has not been reported in humans, tubular abnormalities including cell atrophy, destruction and regeneration as well as interstitial nephritis have been found in hypokalemic patients [12], and these may contribute to the development of renal cysts.
Increased production of intrarenal ammonia secondarily because of hypokalemia is another possible explanation of cyst formation in the context of chronic hypokalemia [4]. The enhanced renal production of ammonia may be linked to cyst formation by a number of mechanisms, including complement activation, stimulating DNA, RNA and protein synthesis and metabolic factors associated with renal ammoniagenesis [13].
Our patient exhibited nephrocalcinosis together with the clinical features of classic Bartter syndrome [2] and a mutation of CLCNKB. Nephrocalcinosis, which is almost invariable in antenatal Bartter syndrome, was previously thought to be absent in classic Bartter syndrome [2] or in Bartter syndrome type III [8]. However, a recent study demonstrated that mutations of CLCNKB could exhibit variable Bartter phenotypes including antenatal Bartter syndrome, classic Bartter syndrome and Gitelman syndrome [9]. Therefore, patients with Bartter syndrome type III may develop nephrocalcinosis. Because nephrocalcinosis can cause nephron obstruction [5], it may also have contributed to the development of renal cysts in our patient. However, the reason why renal cysts are not exhibited by patients with antenatal Bartter syndrome and nephrocalcinosis or classic Bartter syndrome remains unclear.
References
Hebert SC (2003) Bartter syndrome. Curr Opin Nephrol Hypertens 12:527–532
Rodríguez-Soriano J (1998) Bartter and related syndromes: the puzzle is almost solved. Pediatr Nephrol 12:315–327
Torres VE, Young WF Jr, Offord KP, Hattery RR (1990) Association of hypokalemia, aldosteronism, and renal cysts. N Engl J Med 322:345–351
Alpern RJ, Toto RD (1990) Hypokalemic nephropathy—a clue to cystogenesis? N Engl J Med 322:398–399
Igarashi T, Shibuya K, Kamoshita S, Higashihara E, Kawato H, Hagishima K, Kosugi T (1991) Renal cyst formation as a complication of primary distal renal tubular acidosis. Nephron 59:75–79
Stewart PM, Edwards CRW (1990) Hypokalemia, aldosteronism and renal cysts. N Engl J Med 323:129–130
Moudgil A, Rodich G, Jordan SC, Kamil ES (2000) Nephrocalcinosis and renal cysts associated with apparent mineralocorticoid excess syndrome. Pediatr Nephrol 15:60–62
Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, Schurman S, Nayir A, Alpay H, Bakkaloglu A, Rodriguez-Soriano J, Morales JM, Sanjad SA, Taylor CM, Pilz D, Brem A, Trachtman H, Griswold W, Richard GA, John E, Lifton RP (1997) Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet 17:171–178
Konrad M, Vollmer M, Lemmink HH, van den Heuvel LPWJ, Jeck N, Vargas-Poussou R, Lakings A, Ruf R, Deschênes G, Antignac C, Guay-Woodford L, Knoers NVAM, Seyberth HW, Feldmann D, Hildebrandt F (2000) Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J Am Soc Nephrol 11:1449–1459
Igarashi T, Kosugi T (1994) The incidence of renal cyst formation in patients with primary distal renal tubular acidosis. Nephron 66:474
Walsh-Reitz MM, Toback FG (1983) Kidney epithelial cell growth is stimulated by lowering extracellular potassium concentration. Am J Physiol 244:C429–C32
Schwasrtz MM (1998) Hypokalemic nephropathy. In: Jennette JC, Olson JL, Schwartz MM, Silva FG (eds) Heptinstall’s pathology of the kidney, 5th edn. Lippincott-Raven, Philadelphia, pp 662–665
Torres VE, Mujwid DK, Wilson DM, Holley KH (1994) Renal cystic disease and ammoniagenesis in Han:SPRD rats. J Am Soc Nephrol 5:1193–1200
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Watanabe, T., Tajima, T. Renal cysts and nephrocalcinosis in a patient with Bartter syndrome type III. Pediatr Nephrol 20, 676–678 (2005). https://doi.org/10.1007/s00467-004-1732-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-004-1732-5