
Abstract
Chemical extraction of chitin is very hazardous and costly which can be overwhelmed by microbial bioprocessing. In this study, potent protease and lactic acid-producing bacteria were screened and identified as Alcaligens faecalis S3 and Bacillus coagulans L2, respectively. Productions of protease and lactic acid by the respective bacterial strains were optimized. The shell of Litopenaeus vannamei was sequentially treated with the partially purified protease and lactic acid and the treatment conditions were optimized for betterment of chitin yield. Spectral characterization by SEM–EDS, IR, XRD, NMR, XPS and thermal characterization by TG and DTG analysis of the extracted chitin was made and compared with commercial one. It was revealed that both the chitin have similar characteristics. Therefore, it can be articulated that chitin can be extracted from crustacean shells in pure form by microbial bioprocessing which will be a good catch for biorefinary industries for chitin extraction through greener route.
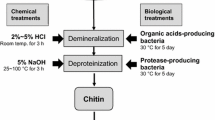
Graphic abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Biomass refinery from renewable bioresources is one of the prime focuses of twenty-first century owing to its potential for sustainable production of value-added materials through waste valorization which is in tune with the principles of circular economy. Bioactive compounds of marine origin are continuously being explored which plays imperative role in human welfare. In the seafood-based industries, about 25–30% of the whole biomass is edible and the rest fractions are considered as waste where the shell part contributes Lion’s share. Worldwide, yearly about 6–8 million tons of crustacean shell is produced and in this context China, India, Indonesia and Thailand are pioneer [1]. Reasonable quantity of chitin is present in crustacean shells (15–30%) along with protein (15–50%) and minerals (30–50%) [2]. Chitin is a linear unit of 2-acetamide-2-deoxy-ß-D-glucopyranose which is linked with ß-(1–4)-linkage, is the second most abundant insoluble carbohydrate in the universe. It is broadly distributed among different arthropods (crustaceans, insects), corals and fungi as a structural component [2,3,4]. Chitin has expanded applications in biotechnology, medicine, pharmacy, cosmetics, textiles, food industry and agriculture for its biodegradability, bio-compatibility and non-toxic nature. Recent studies reveal that its deacetylated derivative chitosan have novel and diversified functional enrollments as preservative, antioxidant, antimicrobial, chelator, emulsifier, fiber, drug carrier, artificial membrane, wound dressing agent and so forth [5,6,7].
For large-scale production of chitin and chitosan, crustacean shell waste is the principal available raw materials. In the shell, chitin are bound with protein and appeared as chitin-protein complex disperse with some minerals (principally calcium), lipids and pigment. For commercial extraction of chitin from the heterogeneous shell, strong acid and alkali are generally employed for demineralization and deproteinization, respectively. Though the use of strong acid and alkali are being practiced by the concerned biorefinary industries, treatment with these non-specific agents may cause partial deacetylation and defragmentation (depolymerization) of the chitin polymer which resulted in inconsistent final product [8, 9]. In the environmental perception, the chemical extraction is not feasible as it leads to potential toxic waste exposure which creates waste disposal problems. To minimize the adverse effect, neutralization and detoxification of the effluent water is necessary prior discharge. Moreover, the nutritive and therapeutic values of the liquid came out after deproteinization could not be addressed due to the presence of strong chemicals. To overcome the shortfalls of chemical deprotenization and demineralization, recently few studies have been undertaken to explore microbes and/or their metabolites to carry out the extraction process which is eco-friendly, safe and economic too. However, the success and costing of the processes are still under question which restrict the commercialization of the process.
Now-a-days, white leg shrimp (Litopenaeus vannamei) is most economical commercially available shrimp all over Asia, and the concern food sectors produces huge amount of shell waste during processing and packaging [10]. Due to lack of comprehensive knowledge about the reclamation aspect, many processing centres do not go for any valorization step and likes to discard the waste. Considering the quantity of the waste, the present study was designed to explore potent protease and organic acid-producing bacterial strain followed by application of the metabolites (protease and organic acid), respectively, for deproteinization and demineralization of shrimp shell for biological extraction of chitin. The optimizations of the metabolite production as well as bio-extraction were also addressed. Additionally, comparative analysis of the physico-chemical features of the extracted chitin and commercial chitin was also made through microscopic, spectrophotometric and thermal analysis.
Materials and methods
Collection of shrimp shell waste
Shrimp shell waste of Litopenaeus vannamei was collected from different areas of West Bengal, India and transported to the laboratory under freezing conditions. Thereafter, the waste was thoroughly washed with water and dehydrated by heating at 60 °C. Then, the shell waste turned into powder (800–1000 µm) by a kitchen grinder.
Bacterial isolation
Isolation of protease-producing bacteria
Soil samples were collected from the shrimp culture ponds and inoculated on media containing (g/L): NaNO3, 2.0; K2HPO4, 1.0; MgSO4.7H2O, 0.5; NaCl, 0.5; skimmed milk powder, 5.0; agar, 20.0 (pH 7.0) through dilution plating technique. After inoculation, the plates were incubated at 35 °C for 48 h. Formation of clear zone around the bacterial colonies indicates proteolysis. The bacterial colony showing higher clear zone/colony size ratio were primarily screened and pure cultured in same media.
Isolation of lactic acid-producing bacteria
For the isolation of lactic acid bacteria soil, pond water and human fecal samples were collected and inoculated on MRS agar media through dilution plating technique. The plates were incubated in anaerobic condition at 35 °C for 24 h. Distinct bacterial colonies were pure cultured and subsequently inoculated in 50 mL of MRS broth. The inoculated media were incubated in anaerobic condition at 35 °C in rotary shaker (120 rpm) and the culture supernatant was collected in day-wise manner for 3 successive days. After centrifugation (5000 rpm, for 10 min), the concentration of lactic acid was measured. The bacteria which produced highest lactic acid was finally selected.
Molecular characterization of bacterial strains
Genomic DNA from the selected bacteria was isolated following the methods of Halder et al. [11] with few modifications. First, the bacterial broth culture was centrifuged at 5000 rpm for 2 min in a micro-centrifuge tube. Then, a solution of 1 M tris HCl (50 µL), 2 N of NaCl (50 µL) and 10% sodium dodecyl sulfate (SDS) were added to the pellet and the volume was adjusted upto 1 mL by MilliQ water and kept for 1.5 h at 50 °C. After that, mixture of chloroform and phenol (1:1, 1 mL) was added and centrifuged at 12,500 rpm for 10 min. The supernatant was taken and mixed with same volume of chloroform and again centrifuged at 13,500 rpm for 10 min. Then, isopropanol and sodium acetate were mixed with the supernatant and kept overnight at − 20 °C. The solution was centrifuged at 14,000 rpm for 10 min. The pellet was re-suspended with 70% ice-cold ethanol and further centrifuge at 14,000 rpm for 10 min. Finally, the pellet (DNA) was dried and dissolved in molecular biology graded water (Himedia). The 16S rRNA gene fragment portion was amplified by using a universal primer (27F- 5′AGAGTTTGATCCTGGCTCAG3′ and 1492R- 5′GGTTACCTTGTTACGACTT3′) [12] under modified PCR condition (30 s for initial denaturation at 94 °C; 30 s for annealing at 50 °C for 30 s; 2-min extension at 72 °C) for 30 cycles. The single band (around 1500 bp) of the PCR product was revealed through 1% agarose gel electrophoresis. The consensus sequence generated after sequencing from forward and reverse sequence data and subsequently analyzed using BLASTn. On the basis of sequence similarity, top ten sequences were retrieved and subjected for phylogenetic analysis using Phylip 3.69. According to the sequence similarity as well as phylogenetic analysis, the taxonomical affiliation of the selected bacterial strains was determined.
Assay of protease activity
Culture supernatant (50 μL) was added to 450 μL 0.2 M phosphate buffer (pH 7.0) containing 1% (w/v) casein and incubated for 1 h at 35 °C. The reaction was terminated by adding 10% trichloroacetic acid (TCA) and kept at room temperature for 15 min. After centrifugation (10,000 rpm for 10 min), the supernatant was collected and free amino acids and soluble peptides are measured by following Lowry’s method [13] using tyrosine (0–10 mg/mL) as standard. One unit protease activity was defined as the amount of enzyme required to liberate 1 μmol of tyrosine (equivalent) per min under standard assay condition.
Estimation of lactic acid
The amount of lactic acid was measured according to Borshchevskaya et al. [14]. Briefly, the centrifuged culture supernatant was diluted 20-fold with distilled water. Thereafter, 50 μL of diluted supernatant was added to test tubes containing 2 ml of iron(III) chloride solution (0.2%). The color generated in the reaction was measured at 390 nm. A calibration curve was prepared following the same procedure by taking different concentration of lactic acid. The final concentration was expressed in g/L.
Optimization of culture conditions of protease and lactic acid production
To find out ideal physico–chemical conditions for protease and lactic acid production by the potent isolates, different parameters and their levels are varied during the course of submerged fermentation. To explore the optimum conditions of protease production, first, different organic and inorganic nitrogenous compounds (viz. casein, peptone, beef extract, yeast extract, NH4Cl, KNO3) are separately supplemented (0.5% w/v) in the production medium. After screening of the best nitrogen source, its concentration was varied between 0.1 and 1.0% (w/v). To find out, ideal carbon sources (viz. glucose, maltose, lactose, fructose, starch) were supplemented separately at 0.5% (w/v) level. After optimization of the best carbon source, the concentration of the same was varied between 0.1 and 1.0% (w/v). The effect of physical parameters like incubation time (12–72 h), pH (5–9), temperature (25–45 °C) on protease production of the selected organism was also determined. The effect of incubation periods (12–48 h) and temperatures (25–45 °C) on lactic acid production by the selected strain were optimized by growing the isolate in MRS broth medium. All the experiments were carried out in 50 mL of the liquid medium in 250-ml Erlenmeyer flasks for the production of both the metabolites and incubated in BOD-shaker at 120 rpm (in anaerobic condition in case of lactic acid production).
Partial purification of protease and lactic acid
For the separation of protein content from fermented mass, 80% ammonium sulfate precipitation of the culture supernatant was carried out. The precipitate was recovered through centrifugation at 10,000 rpm for 15 min. In 50-mM phosphate buffer (pH 7.0), the precipitate was dissolved, dialyzed and the dialysate was considered as crude protease preparation [15]. For lactic acid, the cell-free supernatant was heated at 80 °C for the deactivation of proteins present in the supernatant. Subsequently, the supernatant was filtered through a 0.25-µM filter.
Deproteinization of shrimp shell waste
The powdered shrimp shell was mixed with crude protease and kept at rotary shaker at 120 rpm. The effect of the enzyme to substrate ratio (2:1, 4:1, 6:1, 8:1, 10:1), time of treatment (1, 2, 3 h), treatment pH (5, 6, 7, 8, 9, 10), treatment temperature (20, 30, 40, 50, 60, 70 °C) were optimized. The enzymatic reaction was terminated by heating at 80 °C for 20 min and then thoroughly washed with water. The protein content in treated shell was measured and compared with the protein content of dried native shell. The dried shell matter (both native and treated) of 0.05 g was mixed with 5% (w/v) 10-mL NaOH and heated at 95 °C for 2.5 h. The supernatant was collected and protein concentration was estimated through Lowry's method [13]. Then, the degree of deproteinization was estimated as percentage using the following equation [16]:
where O and R are the mass (g) of the original sample and treated residue in dry weight basis; while, PO and PR are protein contents (g/g) before and after treated, respectively.
Demineralization of deproteinized shell
After deproteinization step, dried shell was mixed with partially purified lactic acid and kept at rotary shaker at 120 rpm. The effect of shell (g) to acid (ml) (0.5:100, 1:100, 2:100, 3: 100, 4: 100 and 5:100), treatment time (15, 30, 45, 60 min) and treated temperature (20, 30, 40, 50, 60, 70 °C) were optimized. After treatment, the solid matter was extracted and washed repeatedly. The measurement of the mineral content after different treatments was done through energy-dispersive X-ray spectroscopic (EDS) analysis (INCA entaFET × 3, Oxford Instrument, UK).
Chitin recover assay
After deproteinization and demineralization step, the final product was treated with distilled water thrice and placed in an oven at 80 °C for dehydration. The chitin content was estimated using the following formula [10]:
Chitin characterization
Scanning electron microscopy (SEM) with energy-dispersive spectroscopy (EDS)
The surface morphology of shrimp shell, treated shell (at each step) and pure chitin were compared by scanning electron microscope (SEM) (Carl ZEISS, SMT, Germany) following gold-coated sputtering method (POLARON-SC7620, POLARON, UK). Simultaneously, the elemental composition in the samples was carried out through energy-dispersive X-ray spectroscopic (EDS) measurements (attached with SEM) (INCA entaFET × 3, Oxford Instrument, UK) attached with SEM.
IR analysis
Infrared spectra were acquired of differently treated samples using IR spectrometer (Perkin Elmer, UK). Sample of 10 mg was scanned at room temperature (25 °C) in the range between 400 and 4000 cm−1. The degree of acetylation (DA) was measured using the following equation [17]:
where A = 1650 cm−1 (amide band) was used as an analytical band and A = 3450 cm−1 (hydroxyl band) was used as the internal reference.
X-ray diffraction analysis
The crystallinity and structural characterization of extracted chitin and pure chitin were studied using an X-ray diffractometer (Bruker, XRD diffractometer, Germany) with CuKα radiation. In the scattering range (2θ) between 5 and 40°, the relative intensity was estimated. The relative crystallinity was measured using Origin 6.0 (Microcal Software INC.) The crystallinity index was determined by the following formula [10]:
where I110 is the maximum intensity (arbitrary unit) of the diffraction (110) at 2θ = 19° and Iam is the intensity of the amorphous diffraction at 2θ = 12.6°.
X-ray photoelectron spectroscopy analysis
The X-ray photoelectron spectroscopies (XPS) of the native and treated shell was carried out and compared with commercial chitin along with the different demineralized condition. Measurements through XPS were executed using a PHI 5000 Versa Probe III spectrometer (Physical Electronics, USA) with an Al Kα X-ray source. The X-ray survey patterns of each sample were recorded in the kinetic energy range of 340–360 eV. The high-resolution spectra of Ca 2p was checked to estimate the detailed chemical composition by deconvolution of their peak components.
Solid-state 13C NMR spectra analysis
The NMR spectra of commercial chitin and extracted chitin were recorded from 0 to 300 ppm by a 500-MHz solid-state 13C CP/MAS NMR (Bruker, AVANCE III, 500-MHz AV 500). The carbon bonds of carbonyl, methyl groups and D-glucopyranosyl ring (C1–C6) was recorded.
Thermal analysis
The thermo-gravimetric analysis (TG) and derivative thermo-gravimetric (DTG) analysis were performed for both commercial and extracted chitin using EXSTAR SII 6300 combine with DTA flexibility. The sample mass around 10 ± 0.50 mg was placed in alumina pan and heated from 30 to 700 °C at 15 °C/ min under the presence of nitrogen gas flowing at 200 ml/min. An alumina powder was taken as reference. From the curves, the temperature at the onset and at the end of the thermal decomposition as well as maximum decomposition of samples was analyzed.
Statistical analysis
Statistical analyses were done using Sigma plot 14.0 software and results were represented as mean ± standard error. MS-Excel and Origin 9.0 were used for calculation and graphical representation of the experimental data.
Results and discussion
Screening of protease and lactic acid-producing bacteria and their molecular characterization
Twenty protease-producing bacteria were primarily screened on the basis of CZ/CS ratio (1.87–5.65) from the soil samples and among them, one potent protease-producing bacterial strains designated as S3 exhibited the highest proteolytic activity. Depending on the result, the strain was selected for further study on the deproteinization of crustacean shells. Simultaneously, 14 lactic acid-producing bacteria were screened and among them the highest lactic acid producer (designated as L2) was selected on the basis of production of lactic acid under submerged fermentation. Therefore, this isolate was subsequently deployed for demineralization of shrimp shells. Taxonomic affiliation of both the bacterial strain was evaluated on the basis of 16S rRNA gene profiling. Through nucleotide homology searching, it was revealed that S3 was 98.3% similar with Alcaligens faecalis and therefore named as A. faecalis S3 (GenBank accession no. MW740143). On the other hand, the lactic acid-producing organism L2 was 99.93% similar with Bacillus coagulans and thus named as B. coagulans L2 (GenBank accession no. MZ031392).
Optimization of protease and lactic acid production
Selection of ideal substrate for maximum production of microbial metabolites is necessary to make any production process proficient. Among the different nitrogenous sources examined, casein at 0.1% (w/v) endorsed maximum protease production (16.44 U/ml) by A. faecalis S3 under submerged fermentation (Fig. 1). Likewise, glucose (0.2% w/v) proved as potential inducible carbon source supported maximum protease production of 28.72 U/ml (Fig. 1). The physical parameters were also playing very important role in any fermentation and in the present study optimum production of protease production was achieved at pH 7.0 and 35 °C temperature after 48 h (Fig. 1). At the end of the OVAT optimization process, 37.40 U/mL of protease production was achieved. Alongside, it was revealed that the highest production of lactic acid by Bacillus coagulans L2 strain was achieved at 35 °C after 24 h of fermentation (Fig. 1) in MRS broth where the initial pH was maintained at 7.0.

Effect of a nitrogen sources, b carbon sources, c concentration of ideal nitrogen and carbon source, d fermentation time, e pH and f temperature on protease production by Alcaligens faecalis S3, and effect of g temperature and h fermentation time on lactic acid production by Bacillus coagulans L2
Enzymatic deproteinization of shrimp shells waste and process optimization
To get chitin from shrimp shell waste, removal of proteins is major and obligatory step. For deproteinization of shrimp waste, the later was treated with the crude protease of A. faecalis S3 at different enzyme/substrate ratio to evaluate efficacy of the protease. The highest percentage of protein removal (82.94%) was obtained at the enzyme to substrate ratio of 1:8 after 3 h of treatment [at unoptimized pH (7.0) and temperature (40 °C)] (Fig. 2). Further increase of the enzyme to substrate ratio results in no significant (P < 0.05) change in degree of deproteinization. When the pH and temperature were addressed, it was found that the degree of deproteinization was optimum at pH 8.0 and at 50 °C (Fig. 2). At the end of the optimization, the maximum removal of protein of 87.77% was achieved. However, complete deproteinization was not achieved in single step/treatment due to various reasons which were pointed out and explained by several researchers. Hajji et al. [17] explained that protease cannot able to liberate the protein which is associated with the chitinous matrix through covalent bonds at C-2 amino groups which act as a shield (Maillard reaction) to obstruct the hydrolysis reaction. While Hamdi et al. [18] reported that the enzymatic activity was failed to take place at the later stage of hydrolysis due to the production of chitin, amino acids and numerous low molecular weights of peptides [19, 20]. Therefore, to obtain maximum deprotenization, the residue was re-suspended in distilled water, washed and further treated with protease with the same conditions. The results revealed that overall, 91.44% of deprotenization was achieved after 1 h of treatment. Comparative account of our finding with other studies revealed that similar deproteinization efficacy was achieved after treatment of shell with Bacillus mojavensis A21 and Bacillus cereus SV1 at crude enzyme to substrate ratio of 10 and 20, respectively [8, 21]. In other studies, the degree of deproteinization of 88, 78, 84 and 63% was achieved through fermentation of shell with Bacillus subtilis Y-108 [22], Pseudomonas aeruginosa K-187 [23], B. subtilis [24], P. aeruginosa F722 [25]. A three-step fermentation of shrimp shell powder sequentially by Serratia marcescens B742, Lactobacillus plantarum ATCC 8014 and Rhizopus japonicus M193 was carried out by Zhang et al. [26] to extract chitin and chitosan. In this context, enzymatic deprotenization was superior and smart technology in crustacean shell biorefinary.

Effect of different process parameters on shell deproteinization with respect to process time: a enzyme to substrate ratio (E/S), b pH, c temperature
Demineralization of deproteinized shell
After deproteinization, demineralization was the next refinery step to remove the minerals associated with chitin. Calcium is the predominant mineral associated with crustacean shell. Results revealed that the pH was gradually increased upto 45 min and no significant change in pH was observed in the reaction mixture thereafter. Along with this, the concentration of lactic acid significantly decreased at 45 min. The residue was recovered, washed (with water) and again treated with lactic acid at the same conditions. However, no significant change in pH and lactic acid concentration was noticed. At 100: 1 (acid: shell) ratio and 40 °C, maximum demineralization was received after 45 min of incubation. The demineralization efficiency was tested through EDS study which revealed that at optimized state 98.7% dimeralization efficiency was achieved. The increase in pH and decrease in lactic acid concentration were probably correlated. This is might be due to the fact that lactic acid is reacting with calcium carbonate to produce calcium lactate thus decreased the lactic acid concentration gradually which correspond to the increase of pH [27]. Earlier report showed that fermentation of shell by Lactobacillus paracasei subsp. tolerans KCTC-3074 [28], Lactobacillus acidophilus SW01 [29], Lactobacillus plantarum ATCC8014 [30] takes 48 h for demineralization, while Brevibacillus parabrevis TKU046 [19] and Pseudomonas aeruginosa F722 [25] removes maximum minerals after 4 and 7 days of fermentation, respectively. However, for demineralization of shrimp waste, direct treatment with lactic acid is much useful and hassle-free as it associated with quick turnover. Moreover, unlike fermentative procedure lactic acid treatment needs no nutrient supplementation.
Chitin recovery assay
After successive steps of deproteinization and demineralization, 25.40% chitin recovery was achieved. The chitin content in shrimp shell is varied species to species and even varied in shell of different body parts. Generally, the overall yield of chitin from shrimp shell depends on the aforesaid factors as well as extraction method and could vary at a range from 5 to 20% [19]. Chitin recovery of 22.12, 22.66 and 23.23%, respectively, from giant tiger shrimp, white leg shrimp and tiger shrimp was reported by Doan et al. [19] following biological method.
Characterization of chitin
SEM–EDS analysis
For assessment of changes in morphology along with mineral composition, shrimp shell in native state and after each step of treatment were examined through SEM–EDS and compared with commercial chitin. In contrast to native shell, after deproteinization, the shells become porous as the proteins dislodged from the rigid shells (Fig. 3a, b). After deproteinization and demineralization, the extracted chitin exhibited the lamellar, microfibrillar arrangement on the surface with less dense packing characteristics of the extracted matter which is comparable with the commercial chitin which showed the uniformly arranged microfibrillar structure (Fig. 3c, d). Similar characteristic lamellar organization of chitin was also noticed by Knidri et al. [31] and Al Sagheer [32] in case of α-chitin.

Scanning electron microscopic views of a native shell, b deproteinized shell, c deproteinized and demineralized shell and d commercial chitin, and corresponding EDS graphs: e native shell, f deproteinized shell, g deproteinized and demineralized shell and h commercial chitin
To examine the dimeralization efficiency during the course of the treatment, elemental composition was also evaluated of the subjected shell at different stage of the treatment procedure and compared with the commercial counterpart (Fig. 2e–h). The elemental analysis revealed that after deproteinization maximum calcium remain associated with the shell and only a fraction of it was dislodged (19.28%) which remained in a complex with shell protein. Eventually, after demineralization, the calcium content reduced upto 98.7% which endorse practicability of the organic acid-mediated demineralization method (Fig. 2g). In earlier report, 96% removal of calcium after treatment with 0.8 M alkali and 0.4 M acid was documented [33]. In contrast, the present study showed better demineralization efficiency. After demineralization, the extracted chitin contains very little amount of calcium along with trace amount of other minerals such as magnesium, ferrous, potassium which is as like as commercial chitin (data not incorporated). The SEM–EDS analysis collectively showed that the chitin present in the heterogeneous shell was extracted sequentially and structural similarity with commercial counterpart.
IR analysis
To check the level of purity, IR spectroscopic analysis of extracted chitin was carried out and compared with commercial one (Fig. 4a). Absence of peaks around 1420–1430 and 1798 cm−1, and only presence of peak around 876 cm−1 of small intensity indicated that both the extracted and commercial chitin has very little amount of calcium carbonate. The spectrum exhibited that both extracted and commercial chitin have characteristic bands around 526, 559, 690, 896, 951, 1009, 1067, 1112, 1154, 1203, 1260, 1308, 1375, 1557, 1621, 1656, 2890, 2932, 2964, 3096, 3257 and 3440 cm−1. In the spectrum, amide I band split into two separated peaks around 1656 and 1621 cm−1 due to hydrogen bonds between intermolecular of –CO.NH and intramolecular of -CO.HOCH2, respectively (which resembles as α-chitin), while amide II band showed at 1557 cm−1 [3, 31]. The bands for the identification of chitin are –OH, –NH and –CH group [17], which are predominant in the IR spectrum around 3440–3447 cm−1, 3265–3266 cm−1 and 2927–2933 cm−1, respectively. Deformation of C-H bond revealed through the band around 1154 cm−1 and 896 cm−1 which exhibited the β-1,4 glycosidic bond of the polysaccharide [34, 35]. For the symmetric and asymmetric vibration of –CH3 and –CH2 group, peaks around 2932 and 2890 cm−1 were apparent in the spectra. The other groups such as amide III, wagging of –CH2, symmetric deformation of –CH3, –CH3 wagging with the side chain, stretching of –CO group are also present in the IR spectra of both the chitin.

Comparison between commercial chitin and extracted chitin a infrared spectrum and b XRD spectrum
In chitin, the repeating N-acetyl-D-glucosamine unit is acetylated at C-2 position while some of the unit stayed free. The degree of acetylation (DA) is an important factor that may affect the properties of chitin like solubility, biodegradability, chemical shifts, etc. The DA is depending on extraction procedure, type of chitin and species variation of shrimp. In this study, the degree of acetylation of commercial chitin (73.46%) and extracted chitin (73.81%) exhibited almost similar percentage of DA (Table 1).
XRD analysis
X-ray diffraction study was carried out to reveal the polymorph structure and crystallinity index (CrI) of chitin. In this study, the comparison between extracted chitin and commercial chitin was carried out through XRD analysis (Fig. 4b). Considerable similar banding pattern with major sharp peaks (2θ) around 9° and 20° in the diffraction planes (020) and (110) represented the orthorhombic crystalline structure, with weak peaks around 13°, 23°, 26°, 35°, 39° in commercial chitin and 23°, 35°, 39° in extracted chitin. These patterns also proved that the extracted chitin has a very resemblance to the α-form of chitin [10]. Perusal of literature advocated that peaks at 9° and 20° are due to the presence of N-acetyl-D-glucosamine and N-glucosamine sequences (respectively) present in chitin chain [35]. It was reported that XRD peaks around 30° and 35° were due to the number of mineral contents and complex compound [Ca10(PO4)6(OH)2] present in the chitin [2]. Results showed that both the commercial and extracted chitin have no peaks at 30° and a weak peak at 35° which revealed that there is a very minimum amount of minerals present in both the matter [10].
Furthermore, the study of XRD patterns revealed that the CrI of extracted chitin was 82.08% which is close with commercial chitin (86.91%) (Table 1). Previous study reported that the extracted chitin from crab (Carcinus mediterraneus) and shrimp (Penaeus kerathurus) had 64.1% and 67.8% CrI, respectively [17]. Marzich et al. [10] reported that CrI of extracted chitin was 67.5% and 67% when shell was treated with trypsin and ficin, respectively. Gbenebor et al. [33] found that CrI value ranges from 79.4 to 87.4% after acid and alkali treatment, and the same is decreases due to loss of N-acetyl groups of the polymer. Therefore, in this study, the extracted chitin showed good crystallinity which represents the stability of chitin structure. Kaya et al. [36] reported that the CrI is varies largely between 43 and 91.7% on the basis of origin.
XPS analysis
X-ray photoelectron spectroscopy (XPS) is a quantitative approach for enumerating the elemental composition of any surface material. In the present study, XPS analysis was performed to access the presence of calcium as elementary component in the shell in native state and after each step of treatment. In the native shells, presence of high peak for Ca2p3/2 and Ca2p1/2 in the spectra attested high calcium content (Fig. 5a). The presence of the minerals is due to the formation of chitin–mineral (salt) complex in the shell of crustacean. After deproteinization, a small amount of calcium was extracted and as a consequence the Ca2p core levels are decreased. Eventually, after treatment with lactic acid, no detectable peak was observed which endorsed that maximum calcium was eliminated (Fig. 5a).

a XPS analysis of shrimp shell on various treatment stages, b 13C solid-state NMR spectrum of commercial chitin (up) and extracted chitin (down)
Solid-state 13C NMR spectra analysis
In this study, the chemical shifts of conformational changes of the extracted and commercial chitin was carried out and the spectra are shown in Fig. 5b. Eight single peaks are consisting of six (C1–C6) spectrums corresponding to the carbon atom of the monomeric unit of N-acetylglucosamine and two of them consist of carbonyl group (C=O) and methyl group (–CH3) of the chitin polymer. All the carbon spectrum appeared between 54 and 106 ppm: 104.6 and 105.04 (C1), 55.20 and 55.11 (C2), 73.61 and 73.41 (C3), 83.53 and 84.60 (C4), 75.89 and 76.22 (C5) and 60.93 and 61.28 (C6) for extracted and commercial chitin, respectively. The strong resonance at 174.1 and 174.07 ppm of carbonyl and 23.17 and 22.98 ppm of a methyl group for extracted and commercial chitin, respectively, was exhibited (Fig. 5b). The clear separated peaks of the C3 and C5 exhibited that the prepared chitin is α-polymorph whose spectral pattern is similar to the commercial chitin [17, 37].
Thermal property analysis
Owing to crystalline polysaccharide nature, degradation of chitin requires high thermal energy. Therefore, the stability under high temperature is essential for the potential applications which can be easily evaluated by TG and DTG. The analyses revealed that there are three stages for the degradation of both extracted and commercial chitin structures (Fig. 6). In the first stage, the loss of weight for both chitin at below 100 °C was noticed which might be due to the moisture or the evaporation of water [38]. In the second stage, the degradation started from 239 °C and maximum loss was achieved before 400 °C. During this stage, 34.9 and 38.6% of in extracted and commercial chitin were degraded, respectively. The degradation might be started from the weak side of C–O–C bond, gradually dehydrate aliphatic compounds (CH2, CH3) from the glucopyranose ring of chitin and subsequently by decomposition and depolymerization of N-acetylated units and amide groups (C=O, N–H) [38,39,40]. In the third stage, the nearly complete denaturation of chitin was observed at 550 °C might be due to the degradation of some organic salts present in the matters [37]. From the DTG curves, it was revealed that highest decomposition temperature (Tmax) was 353 and 337 °C for extracted chitin and commercial chitin, respectively (Fig. 6). The disintegration temperature of chitin from the various aquatic invertebrates (viz. krill, snail, etc.) varies approximately between 350 and 400 °C [41]. Differential thermal analysis (DTA) showed the presence of exothermic peaks due to the cleavage of the glycosidic bond at 372 and 353 °C for the extracted and commercial chitin, respectively. These findings attested that the extracted chitin has comparatively higher thermal stability as compared to the commercial chitin which might be due to adverse effect of chemical extraction process on the structure of commercial chitin.

Thermo-gravimetric a and derivative thermo-gravimetry curve b of extracted chitin (red) and commercial chitin (black)
Conclusion
In the current study, successful chitin extraction from shell of Litopenaeus vannamei was accomplished through the treatment of protease and lactic acid of Alcaligens faecalis S3 and Bacilus coagulans L2, respectively. High deprotenization and demineralization efficiency of the executed extraction process validates the process proficiency. Both the metabolites are produced in simple media and the production process was optimized. Morphological and spectroscopic characterization attested superimposable characteristics of both extracted and commercial chitin. Thermal stability of extracted chitin is higher than the commercial one. The overall extraction process was very efficient for chitin extraction on a short basis of time with environmental acceptability. However, shells of different crustacean origin should be tested for extraction of chitin which will enable broad spectrum application of the extraction process. Moreover, the entire process should be implemented in chitin biorefinary process in large scale in a greener environmentally friendly route.
References
Cheba BA, Zaghloul TI, EL-Mahdy AR (2018) Demineralized crab and shrimp shell powder: cost effective medium for bacillus Sp. R2 growth and chitinase production. Procedia Manuf 22:413–419
Kumari S, Annamareddy SKH, Abanti S, Rath PK (2017) Physicochemical properties and characterization of chitosan synthesized from fish scales, crab and shrimp shells. Int J Biol Macromol 104:1697–1705
Mohammed MH, Williams PA, Tverezovskaya O (2013) Extraction of chitin from prawn shells and conversion to low molecular mass chitosan. Food Hydrocolloids 31(2):166–171
Halder SK, Mondal KC (2018) Microbial valorization of chitinous bioresources for chitin extraction and production of chito-oligomers and N-acetylglucosamine: trends, perspectives and prospects, In Microbial Biotechnolog, Springer, Singapore, pp 69–107
Jayakumar R, Prabaharan M, Kumar PS, Nair SV, Tamura H (2011) Biomaterials based on chitin and chitosan in wound dressing applications. Biotechnol Adv 29(3):322–337
Muxika A, Etxabide A, Uranga J, Guerrero P, De La Caba K (2017) Chitosan as a bioactive polymer: processing, properties and applications. Int J Biol Macromol 105:1358–1368. https://doi.org/10.1016/j.ijbiomac.2017.07.087
Jiang Y, Fu C, Wu S, Liu G, Guo J, Su Z (2017) Determination of the deacetylation degree of chitooligosaccharides. Mar Drugs 15(11):332
Younes I, Ghorbel-Bellaaj O, Nasri R, Chaabouni M, Rinaudo M, Nasri M (2012) Chitin and chitosan preparation from shrimp shells using optimized enzymatic deproteinization. Process Biochem 47(12):2032–2039
Younes I, Hajji S, Rinaudo M, Chaabouni M, Jellouli K, Nasri M (2016) Optimization of proteins and minerals removal from shrimp shells to produce highly acetylated chitin. Int J Biol Macromol 84:246–253
Marzieh MN, Zahra F, Tahereh E, Sara KN (2019) Comparison of the physicochemical and structural characteristics of enzymatic produced chitin and commercial chitin. Int J Biol Macromol 139:270–276
Halder SK, Maity C, Jana A, Pati BR, Mondal KC (2012) Chitinolytic enzymes from the newly isolated Aeromonas hydrophila SBK1: study of the mosquitocidal activity. Biocontrol 57(3):441–449
Chen YL, Lee CC, Lin YL, Yin KM, Ho CL, Liu T (2015) Obtaining long 16S rDNA sequences using multiple primers and its application on dioxin-containing samples. BMC Bioinform 16(18):1–11
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the folin phenol reagent. J Biol Chem 193:65–275
Borshchevskaya LN, Gordeeva TL, Kalinina AN, Sineokii SP (2016) Spectrophotometric determination of lactic acid. J Anal Chem 71(8):755–758
Halder SK, Maity C, Jana A, Das A, Paul T, Mohapatra PKD, Pati BR, Mondal KC (2013) Proficient biodegradation of shrimp shell waste by Aeromonas hydrophila SBK1 for the concomitant production of antifungal chitinase and antioxidant chitosaccharides. Int Biodeter Biodegr 79:88–97
Sedaghat F, Yousefzadi M, Toiserkani H, Najafipour S (2017) Bioconversion of shrimp waste Penaeus merguiensis using lactic acid fermentation: an alternative procedure for chemical extraction of chitin and chitosan. Int J Biol Macromol 1(104):883–888
Hajji S, Younes I, Ghorbel-Bellaaj O, Hajji R, Rinaudo M, Nasri M, Jellouli K (2014) Structural differences between chitin and chitosan extracted from three different marine sources. Int J Biol Macromol 65:298–306
Hamdi M, Hammami A, Hajji S, Jridi M, Nasri M, Nasri R (2017) Chitin extraction from blue crab (Portunus segnis) and shrimp (Penaeus kerathurus) shells using digestive alkaline proteases from P. segnis viscera. Int J Biol Macromol 101:455–463
Doan CT, Tran TN, Vo TP, Nguyen AD, Wang SL (2019) Chitin extraction from shrimp waste by liquid fermentation using an alkaline protease-producing strain, Brevibacillus parabrevis. Int J Biol Macromol 131:706–715
Paul T, Halder SK, Das A, Ghosh K, Mandal A, Payra P, Barman P, Mohapatra PK, Pati BR, Mondal KC (2015) Production of chitin and bioactive materials from Black tiger shrimp (Penaeus monodon) shell waste by the treatment of bacterial protease cocktail. 3 Biotech 5(4):483–493
Manni L, Ghorbel-Bellaaj O, Jellouli K, Younes I, Nasri M (2010) Extraction and characterization of chitin, chitosan, and protein hydrolysates prepared from shrimp waste by treatment with crude protease from Bacillus cereus SV1. Appl Biochem Microbiol 162(2):345–357
Yang JK, Shih L, Tzeng YM, Wang SL (2000) Production and purification of protease from a Bacillus subtilis that can deproteinize crustacean wastes. Enzyme Microb Technol 26(5–6):406–413
Oh YS, Shih L, Tzeng YM, Wang SL (2000) Protease produced by Pseudomonas aeruginosa K-187 and its application in the deproteinization of shrimp and crab shell wastes. Enzyme Microb Technol 27(1–2):3–10
Sini TK, Santhosh S, Mathew PT (2007) Study on the production of chitin and chitosan from shrimp shell by using Bacillus subtilis fermentation. Carbohydr Res 342(16):2423–2429
Oh KT, Kim YJ, Jung WJ, Park RD (2007) Demineralization of crab shell waste by Pseudomonas aeruginosa F722. Process Biochem 42(7):1069–1074
Zhang H, Yun S, Song L, Zhang Y, Zhao Y (2017) The preparation and characterization of chitin and chitosan under large-scale submerged fermentation level using shrimp by-products as substrate. Int J Biol Macromol 96:334–339
Yang PB, Tian Y, Wang Q, Cong W (2015) Effect of different types of calcium carbonate on the lactic acid fermentation performance of Lactobacillus lactis. Biochem Eng J 98:38–46
Jung WJ, Kuk JH, Kim KY, Park RD (2005) Demineralization of red crab shell waste by lactic acid fermentation. Appl Microbiol Biotechnol 67(6):851–854
Duan S, Li L, Zhuang Z, Wu W, Hong S, Zhou J (2012) Improved production of chitin from shrimp waste by fermentation with epiphytic lactic acid bacteria. Carbohydr Polym 89(4):1283–1288
Zhang H, Jin Y, Deng Y, Wang D, Zhao Y (2012) Production of chitin from shrimp shell powders using Serratia marcescens B742 and Lactobacillus plantarum ATCC 8014 successive two-step fermentation. Carbohydr Res 362:13–20
El Knidri H, El Khalfaouy R, Laajeb A, Addaou A, Lahsini A (2016) Eco-friendly extraction and characterization of chitin and chitosan from the shrimp shell waste via microwave irradiation. Process Saf Environ Prot 104:395–405
Al Sagheer FA, Al-Sughayer MA, Muslim S, Elsabee MZ (2009) Extraction and characterization of chitin and chitosan from marine sources in Arabian Gulf. Carbohydr Polym 77(2):410–419
Gbenebor OP, Adeosun SO, Lawal GI, Jun S, Olaleye SA (2017) Acetylation, crystalline and morphological properties of structural polysaccharide from shrimp exoskeleton. Int J Eng Sci Technol 20(3):1155–1165
Mhamdi S, Ktari N, Hajji S, Nasri M, Kamoun AS (2017) Alkaline proteases from a newly isolated Micromonospora chaiyaphumensis S103: characterization and application as a detergent additive and for chitin extraction from shrimp shell waste. Int J Biol Macromol 94:415–422
Moussout H, Ahlafi H, Aazza M, Bourakhouadar M (2016) Kinetics and mechanism of the thermal degradation of biopolymers chitin and chitosan using thermogravimetric analysis. Polym Degrad Stab 130:1–9
Kaya M, Lelešius E, Nagrockaitė R, Sargin I, Arslan G, Mol A, Baran T, Can E, Bitim B (2015) Differentiations of chitin content and surface morphologies of chitins extracted from male and female grasshopper species. PLoS ONE 10(1):e0115531
Bernabé P, Becherán L, Cabrera-Barjas G, Nesic A, Alburquenque C, Tapia CV, Taboada E, Alderete J, De Los RP (2020) Chilean crab (Aegla cholchol) as a new source of chitin and chitosan with antifungal properties against Candida spp. Int J Biol Macromol 15(149):962–975
Juárez-De La Rosa BA, May-Crespo J, Quintana-Owen P, Gónzalez-Gómez WS, Yañez-Limón JM, Alvarado-Gil JJ (2015) Thermal analysis and structural characterization of chitinous exoskeleton from two marine invertebrates. Thermochim Acta 20(610):16–22
Hou F, Ma X, Fan L, Wang D, Ding T, Ye X, Liu D (2020) Enhancement of chitin suspension hydrolysis by a combination of ultrasound and chitinase. Carbohydr Polym 231:115669. https://doi.org/10.1016/j.carbpol.2019.115669
Purkayastha D, Sarkar S (2019) Physicochemical structure analysis of chitin extracted from pupa exuviae and dead imago of wild black soldier fly (Hermetia illucens). J Polym Environ 27:1–3
Liu Y, Xing R, Yang H, Liu S, Qin Y, Li K, Yu H, Li P (2020) Chitin extraction from shrimp (Litopenaeus vannamei) shells by successive two-step fermentation with Lactobacillus rhamnoides and Bacillus amyloliquefaciens. Int J Biol Macromol 148:424–433
Funding
The authors are grateful to the Swami Vivekananda Merit Cum Means Scholarship Scheme and Department of Science & Technology and Biotechnology, Govt. of West Bengal, India for financial assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There is no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rakshit, S., Mondal, S., Pal, K. et al. Extraction of chitin from Litopenaeus vannamei shell and its subsequent characterization: an approach of waste valorization through microbial bioprocessing. Bioprocess Biosyst Eng 44, 1943–1956 (2021). https://doi.org/10.1007/s00449-021-02574-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00449-021-02574-y