
Abstract
Glycerol can be used as a primary carbon source by yeasts, little is known regarding glycerol metabolism in Candida tropicalis. In this study, glycerol kinase gene (gk) was disrupted from xylitol dehydrogenase gene (XYL2) knockout C. tropicalis strain BSXDH-3. The resultant gk knockout C. tropicalis strain was incapable to grow on glycerol. The cells growth on glycerol was resumed by co-expressing Scheffersomyces stipitis gcy1, 2 and 3 genes, which respectively encode NADP+-dependent glycerol dehydrogenase 1, 2 and 3, under the control of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) promoter. NADPH-dependent xylitol production was higher in the engineered strain, termed “GK”, than in BSXDH-3. In fermentation experiments using glycerol as co-substrate with xylose, strain GK produced xylitol 0.85 and 1.28 g l−1 h−1 at the time periods of 16 and 24 h, respectively, which is 30 and 18 % higher at same time intervals in BSXDH-3. This is the first report of gk gene disruption and co-expression of gcy1, 2 and 3 genes for NADPH regeneration and enhanced xylitol production in C. tropicalis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Xylitol is a five-carbon sugar alcohol that has the same order of sweetness like conventional sugar [6]. Xylitol does not need insulin for its metabolic regulation, so it has been used as a sweetener in low energy and diabetics’ foods. Xylitol is used in syrups, tonics, and vitamin formulations [1]. At present, the industrial production of xylitol is based on xylose dehydrogenation in a nickel-catalyzed process which requires high temperature, high pressure and extensive purification steps that are very costly and energy intensive. Therefore, some alternative biological processes using xylose-assimilating microorganisms have been extensively studied, especially those involving yeasts from Candida genus [4, 5, 11, 17]. Considering the disadvantages of the chemical method, xylitol can be produced by C. tropicalis as an alternative and environmentally safe method. The increasing demand for xylitol in the pharmaceutical and food industries has led to interesting efforts in optimizing the biological production of xylitol [7, 9, 19, 20, 23].
In our previous research, XYL2 gene encoding XDH was disrupted in C. tropicalis ATCC20913. The resulting strain BSXDH-3 produced xylitol with a yield of 97 % under fully aerobic condition [12]. Glucose is an inadequate co-substrate because of its repression effect on xylose reductase (XR) which catalyzes the reduction of d-xylose to xylitol. Consequently, glycerol was found to be the best co-substrate for cell maintenance and NADPH regeneration by screening of various carbon sources [13].
Glycerol is used as co-substrate by C. tropicalis for cofactor regeneration and to enhance reducing power of the cell. Glycerol, due to its role in osmoregulation, its metabolic pathways for production and consumption, as well as its mechanisms for intracellular accumulation has attracted increasing attention during the last few years. The use of modern tools in yeast genetics and molecular biology has resulted in many new discoveries concerning both response to osmotic stress and glycerol metabolism in yeasts [16, 18]. The knowledge derived from glycerol metabolism, transport and regulation processes has a strong technological potential for industry and agriculture applications, putatively providing a major breakthrough for interesting biotechnological applications, like tools for the genetic engineering of other organisms [21].
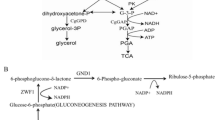
Two pathways of glycerol metabolism were found in yeasts (Fig. 1). The more extensively studied phosphorylative pathway is involving glycerol kinase [2, 3, 14, 15] and the oxidative pathway involving NADP-linked glycerol dehydrogenase [24]. Glycerol is used as a primary carbon source by yeasts, and catabolized by the phosphorylative pathway in C. tropicalis. In this study, we genetically disrupted the phosphorylative pathway and introduced the alternative oxidative pathway to improve the NADPH production. Glycerol kinase gene was disrupted from the genome of xylitol dehydrogenase gene (XYL2)-disrupted C. tropicalis strain BSXDH-3. The resultant gk knockout C. tropicalis strain was incapable to grow on glycerol. The cells growth on glycerol was resumed by co-expressing Scheffersomyces stipitis gcy1, 2 and 3 genes, which respectively encode NADP+-dependent glycerol dehydrogenase 1, 2 and 3, under the control of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) promoter. NADPH-dependent xylitol production was higher in the engineered strain, termed “GK”, than in BSXDH-3.

Schematic summary of glycerol metabolic pathway in C. tropicalis
Materials and methods
Strains
Candida tropicalis L10 (ura3/ura3), a uracil auxotroph derived from C. tropicalis BSXDH-3, was used as a host strain for transformation [13]. Escherichia coli DH5α was used for plasmids preparation.
Culture media
Cultures were grown in YM medium containing 3 g yeast extract l−1, 3 g malt extract l−1, 5 g bactopeptone l−1 and 20 g glucose l−1. For genetic manipulation with C. tropicalis, YNB medium containing 6.7 g yeast nitrogen base without amino acids l−1 and 20 g glucose l−1 and YNB-5FOA medium containing 6.7 g yeast nitrogen base without amino acids l−1, 20 g glucose l−1, 0.1 g Uracil l−1, 0.1 g uridine l−1 and 0.8 g 5-fluroorotic acid l−1 were used. Luria–Bertani (LB) medium was used to cultivate E. coli.
Cloning of gcy1, 2 and 3 genes from S. stipitis
gcy1, 2 and 3 genes encoding GDH1, 2 and 3 were isolated from genomic DNA of S. stipitis (CBS 6054) by PCR, using primers synthesized complementary to the coding regions of genes by an XHL PCR Kit (Bioneer, Daejeon, Korea). The forward and reverse primer sequences used for gene amplification were designed based on reported sequences of gcy1, 2 and 3 genes in S. stipitis genome [8].
Enzyme activity assays
GDH1, 2 and 3 activities were measured by the procedure [10], measuring the conversion of NADP to NADPH as change in A 340 nm. Activities are expressed as specific activity [units (mg of protein)−1], where one unit corresponds to the conversion of 1 μmol of NADP+ per min.
Construction of the disruption cassettes for gk gene of C. tropicalis
Four disruption cassettes were constructed to disrupt gk gene encoding glycerol kinase from C. tropicalis genome. The URA3 gene was amplified from the genomic DNA of C. tropicalis ATCC 20336 by PCR, using primers synthesized complementary to the coding region of URA3 sequence (GenBank Accession no. AB006207). The URA3 gene was inserted into pGEM-T easy vector (Promega) and the resulting plasmid was designated pGEM-URA3. For pop-out of URA3 selection marker via DNA recombination, four DNA sequences (glu, arg, leu and trp) isolated from Bacillus subtilis genome were used. Identical genes were inserted into BglII and BamHI sites on either side of URA3 in pGEM-URA3. The four resulting plasmids contained glu-URA3-glu, arg-URA3-arg, leu-URA3-leu and trp-URA3-trp cassettes, respectively. The cassettes were then inserted into four pairs of gk fragments for integration into C. tropicalis genome by homologous recombination system using lithium acetate method [13]. The constructed cassettes are shown schematically (Fig. 2).

Schematic representation of the strategy used for construction of C. tropicalis gk disruption cassettes. Four copies of gk were disrupted by inserting gk1, gk2, gk3, and gk4 disruption cassettes into C. tropicalis genome
Fermentation conditions and analytical methods
The fermentation medium for xylitol production consisted of 50 g d-xylose l−1, 10 g yeast extract l−1, 5 g KH2PO4 l−1, 0.2 g MgSO4.7H2O l−1 and 20 g glycerol l−1 as a co-substrate for cell growth. Xylitol production was performed in 250 ml Erlenmeyer flask with 50 ml xylitol fermentation medium at 200 rpm in a shaking incubator at 30 °C. The concentration of d-xylose, xylitol, and glycerol were analyzed by HPLC equipped with a Sugar-Pak 1 column (Waters, MA, USA) and a refractive-index detector (Waters). Distilled water was used as the mobile phase at a column temperature of 90 °C and a flow rate of 0.5 ml min−1. Cell growth was monitored spectrophotometrically at 600 nm. One A 600 was equivalent to 0.474 g dry cell weight l−1.
Results and discussion
Purification of recombinant GDH1, 2 and 3
The recombinant GDH1, 2 and 3 were purified from E. coli BL21. The enzymatic activities of GDH1, 2 and 3 are shown in Table 1.
Physiological confirmation of gk gene disruption
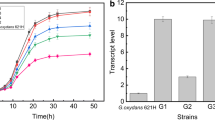
For physiological confirmation of gk gene disruption, parent C. tropicalis and gk knockout C. tropicalis strains were incubated for 3 days on minimal essential medium with glycerol as a sole carbon source for cell growth and maintenance. The parent C. tropicalis strain successfully grew on glycerol minimal essential medium plates. However, gk knockout C. tropicalis strain was incapable of growth on glycerol minimal essential medium plates (Fig. 3a). Our result shows that C. tropicalis is assimilating glycerol for cell growth by phosphorylative pathway involving glycerol kinase and the result obtained coincided with the previous report [22].

Physiological confirmation of gk disruption. The strains were incubated on the minimal essential medium with glycerol as a sole carbon and energy source. a Strains are indicated as: 1 C. tropicalis BSXDH-3, 2 gk disrupted C. tropicalis. b Strains are indicated as: 1 C. tropicalis BSXDH-3, 2 C. tropicalis GK (co-expression of gcy1, 2 and 3)
The oxidative pathway of S. stipitis involving gcy1, 2 and 3 genes encoding GDH1, 2 and 3 is responsible for cofactor regeneration. gk knockout C. tropicalis strain was enabled to assimilate glycerol for cell growth and generation of reducing power by engineering the oxidative pathway of S. stipitis (Fig. 3b).
Construction of gcy1, 2 and 3 co-expression cassettes and yeast transformation
gk disrupted C. tropicalis strain was grown on YNB medium and then spread on YNB-5FOA plates. A URA3 pop-out mutant was selected among the 5FOA-resistant colonies and was used for transformation of first expression cassette. The first constructed cassette is composed of 5′ leu-L1, GAPDH promoter (PGAPDH), gcy1, GAPDH terminater (TGAPDH), tryptophan (trp), uracil marker (URA3), tryptophan (trp) and 3′ leu-R1. The cassette contains two DNA fragments 5′ leu-L1 and 3′ leu-R1 flanking the first target leu region (inserted with gk2 disruption cassette) is transformed into URA3 pop-out mutant by homologous recombination system using lithium acetate method [13]. A similar procedure as adopted for transformation of gcy1 gene expression cassette was used for making the second and third co-expression cassettes containing gcy2 and 3 genes. The genes leu, trp, arg, and glu encoding leucine, tryptophan, arginine, and glutamic acid were cloned from B. substillus genome. Physical map of the co-expressing cassettes and integration of the cassettes into gk disrupted C. tropicalis strain is shown (Fig. 4).

Schematic representation of the strategy used for construction of S. stipitis gcy1, 2 and 3 co-expression cassettes for integration into C. tropicalis genome. gcy1 and gcy2 genes were integrated at leu fragment (inserted with gk2 disruption cassette). gcy3 gene was inserted at hisG fragment (existed in host strain)
Comparison of xylitol production in parent strain BSXDH-3 versus recombinant GK
To evaluate and compare rates of xylitol production in BSXDH-3 and GK, fermentation experiments were performed in 250 ml Erlenmeyer flasks with 50 ml xylitol fermentation medium in a shaking incubator, 200 rpm at 30 °C. Glycerol consumption rate showed no significant difference between BSXDH-3 and PP. Rates of xylitol production at 16 and 24 h for GK were 0.74 and 1.41 g l−1 h−1, respectively; these values are 30 and 18 % higher than corresponding rates for BSXDH-3 (Fig. 5). This strategy for strain development will contribute greatly to other microbiologists who are interested in improving the metabolites production in microorganisms.

Comparison of fermentation profiles of C. tropicalis strains BSXDH-3 (parent) and GK (over-expressing GDH1, 2 and 3). Black and white symbols are used, respectively, for BSXDH-3 and GK, for d-xylose consumption (filled circle, open circle), xylitol production (filled triangle, open triangle), glycerol consumption (filled square, open square) and dry cell weight (closed diamond, open diamond)
References
Brunzell JD (1978) Use of fructose, sorbitol or xylitol as a sweetener in diabetes Mellitus. J Am Diet Assoc 73:499–506
Courtright JB (1975a) Intracellular localization and properties of glycerokinase and glycerophosphate dehydrogenase in Neurospora crassa. Arch Biochem Biophys 167:21–33
Courtright JB (1975b) Differential rates of synthesis of glycerokinase and glycerophospate dehydrogenase in Neurospora crassa during induction. Arch Biochem Biophys 167:34–44
Granström TB, Izumori K, Leisola M (2007) A rare sugar xylitol. Part I: the biochemistryand biosynthesis of xylitol. Appl Microbiol Biotechnol 74:277–281
Granström TB, Izumori K, Leisola M (2007) A rare sugar xylitol. Part II: biotechnological production and future applications of xylitol. Appl Microbiol Biotechnol 74:273–276
Heikkilä H, Nurmi J, Rahkila L, Töyrylä M (1992) Method for the production of xylitol. US Patent 5,081,026
Jeffries TW, Jin YS (2004) Metabolic engineering for improved fermentation of pentoses by yeasts. Appl Microbiol Biotechnol 63:495–509
Jeffries TW, Grigoriev IV, Grimwood J, Laplaza JM, Aerts A, Salamov A, Schmutz J, Lindquist E, Dehal P, Shapiro H, Jin YS, Passoth V, Richardson PM (2007) Genome sequence of the lignocellulose-bioconverting and xylose-fermenting yeast Pichia stipitis. Nat Biotechnol 25(3):319–326
Jin YS, Jeffries TW (2003) Changing flux of xylose metabolites by altering expression of xylose reductase and xylitol dehydrogenase in recombinant Saccharomyces cerevisiae. Appl Biochem Biotechnol 105:277–285
Kikawa Y, Shin YS, Inuzuka M, Zammarchi E, Mayumi M (2002) Diagnosis of fructose-1,6-bisphosphatase deficiency using cultured lymphocyte fraction: a secure and noninvasive alternative to liver biopsy. J Inherit Metab Dis 25:41–46
Kim JH, Han KC, Koh YH, Ryu YW, Seo JH (2002) Optimization of fed-batch fermentation for xylitol production by Candida tropicalis. J Ind Microbiol Biotechnol 29:16–19
Ko BS, Rhee CH, Kim JH (2006) Enhancement of xylitol productivity and yield using a xylitol dehydrogenase gene-disrupted mutant of Candida tropicalis under fully aerobic conditions. Biotechnol Lett 28:1159–1162
Ko BS, Kim J, Kim JH (2006) Production of xylitol from d-Xylose by a xylitol dehydrogenase gene-disrupted mutant of Candida tropicalis. Appl Environ Microbiol 72:4207–4213
North MJ (1973) Cold-induced increase of glycerol kinase in Neurospora crassa. FEBS Lett 35:67–70
North MJ (1974) Cold-induced increase of glycerol kinase in Neurospora crassa: rapid inactivation of the enzyme in vivo. J Bacteriol 120:741–747
Pavlik P, Simon M, Schuster T, Ruis H (1993) The glycerol kinase (GUT1) gene of Saccharomyces cerevisiae: cloning and characterization. Curr Genet 24:21–25
Prakasham RS, Rao RS, Hobbs PJ (2009) Current trends in biotechnology production of xylitol and future prospects. Curr Trends Biotechnol Pharm 3:8–36
Roënnow B, Kielland-Brandt MC (1993) GUT2, a gene for mitochondrial glycerol 3-phosphate dehydrogenase of Saccharomyces cerevisiae. Yeast 9:1121–1130
Sampaio FC, Chaves-Alves VM, Converti A, Lopes Passos FM, Cavalcante Cohelo JL (2008) Influences of cultivation conditions on xylose-to-xylitol bioconversion by a new isolate of Debaryomyces hanseii. Biores Technol 99:502–508
Sánchez S, Bravo V, Moya JJ, Castro E, Camacho F (2004) Influence of temperature on the fermentation of d-xylose by Pachisolen tannophilus to produce ethanol and xylitol. Process Biochem 39:637–679
Serrano R (1996) Salt tolerance in plants and microorganisms: toxicity targets and defense responses. Int Rev Cytol 165:1–52
Sherwood KE, Cano DJ, Maupin-Furlow JA (2009) Glycerol-mediated repression of glucose metabolism and glycerol kinase as the sole route of glycerol catabolism in the haloarchaeon Haloferax volcanni. J Bacteriol 191(13):4307–4315
Silva CJSM, Mussatto SI, Roberto IC (2006) Study of xylitol production by Candida guilliermondii on a bench reactor. J Food Eng 75:115–119
Viswanath-Reddy M, Bennett SN, Howe HB (1977) Characterization of glycerol nonutilizing protoperithecial mutants of Neurospora. Mol Gen Genet 153:29–38
Acknowledgments
This work was supported by the 21C Frontier Microbial Genomics and Applications Center Program, Ministry of Education, Science and Technology, Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ahmad, I., Shim, W.Y. & Kim, JH. Enhancement of xylitol production in glycerol kinase disrupted Candida tropicalis by co-expression of three genes involved in glycerol metabolic pathway. Bioprocess Biosyst Eng 36, 1279–1284 (2013). https://doi.org/10.1007/s00449-012-0872-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00449-012-0872-4