
Abstract
Global emissions of atmospheric CO2 and tropospheric O3 are rising and expected to impact large areas of the Earth’s forests. While CO2 stimulates net primary production, O3 reduces photosynthesis, altering plant C allocation and reducing ecosystem C storage. The effects of multiple air pollutants can alter belowground C allocation, leading to changes in the partial pressure of CO2 (pCO2) in the soil , chemistry of dissolved inorganic carbonate (DIC) and the rate of mineral weathering. As this system represents a linkage between the long- and short-term C cycles and sequestration of atmospheric CO2, changes in atmospheric chemistry that affect net primary production may alter the fate of C in these ecosystems. To date, little is known about the combined effects of elevated CO2 and O3 on the inorganic C cycle in forest systems. Free air CO2 and O3 enrichment (FACE) technology was used at the Aspen FACE project in Rhinelander, Wisconsin to understand how elevated atmospheric CO2 and O3 interact to alter pCO2 and DIC concentrations in the soil. Ambient and elevated CO2 levels were 360±16 and 542±81 μl l−1, respectively; ambient and elevated O3 levels were 33±14 and 49±24 nl l−1, respectively. Measured concentrations of soil CO2 and calculated concentrations of DIC increased over the growing season by 14 and 22%, respectively, under elevated atmospheric CO2 and were unaffected by elevated tropospheric O3. The increased concentration of DIC altered inorganic carbonate chemistry by increasing system total alkalinity by 210%, likely due to enhanced chemical weathering. The study also demonstrated the close coupling between the seasonal δ13C of soil pCO2 and DIC, as a mixing model showed that new atmospheric CO2 accounted for approximately 90% of the C leaving the system as DIC. This study illustrates the potential of using stable isotopic techniques and FACE technology to examine long- and short-term ecosystem C sequestration.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Physiological effects of elevated atmospheric CO2 and tropospheric O3
Global CO2 emissions have risen consistently from pre-industrial levels of 275 μl l−1 to current levels in excess of 360 μl l−1, with a concentration of 550 μl l−1 predicted by the middle of this century (Houghton et al. 1996). Concurrent with increasing atmospheric CO2, tropospheric O3 levels have risen from pre-industrial levels of 10 nl l−1 to current summer daytime O3 levels of 50–70 nl l−1. Estimates of increases in tropospheric O3 of 1–2% per year may result in a tripling of O3 concentrations within the next 30–40 years (Chameides et al. 1994; Marenco et al. 1994). Even at low concentrations plants exhibit physiological toxicity to O3 (Karnosky et al. 1996). With 50% of the world’s forests expected to experience increased exposure to both CO2 and O3 by 2100 (Fowler et al. 1999), the resulting balance of stimulatory/inhibitory effects on physiological processes may profoundly influence the C stocks of terrestrial ecosystems.
The interacting effects of these two atmospheric pollutants on ecosystem C dynamics are largely unknown, although their individual effects have been well studied. C3 plant photosynthesis is limited by the concentration of atmospheric CO2, and an increase in the ratio of CO2:O2 will increase C assimilation (Tolbert and Zelitch 1983; Sage 1994). This increase in net photosynthesis under elevated CO2 has consistently been found to change C cycling in forest ecosystems, including: (1) greater C inputs to soil due to increased rates of litterfall, root turnover and rhizodeposition (Pregitzer et al. 1995; DeLucia et al. 1999); (2) increased fungal and bacterial activity and enhanced substrate degradation (Phillips et al. 2002); and (3) increased partial pressure of soil CO2 (soil pCO2) (Andrews and Schlesinger 2001; King et al. 2001).
Whereas CO2 stimulates photosynthesis, increased tropospheric O3 reduces photosynthesis compared to ambient concentrations (Reich et al. 1990). The decrease in photosynthesis is caused by oxidative damage to cell membranes, light harvesting processes, C fixation, and disruption of C allocation patterns (Pell 1987; Landry and Pell 1993; Chappelka and Samuelson 1998). Oxidative damage can lead to a number of negative effects on forest productivity that ultimately decrease ecosystem C accumulation. These effects include decreased stomatal conductance and photosynthesis, premature leaf abscission, and decreased leaf size (Coleman et al. 1995). The interactive effects of elevated CO2 and O3 have recently been found to strongly influence ecosystem C storage. Loya et al. (2003) found that when forest plots under elevated CO2 are also exposed to elevated O3, they formed less new soil C than plots exposed to elevated CO2 alone. However, virtually nothing is known about how elevated CO2 and O3 will interact to affect the inorganic C chemistry of soil solutions in forested ecosystems.
Soil pCO2-dissolved inorganic carbonate interactions
CO2 and O3 alter leaf-level plant dynamics, affecting the allocation of photosynthate to root systems, quality of litter inputs, and soil respiration. As CO2 accumulates in the soil due to the respiration of plant roots and associated soil microorganisms, concentrations of up to 100x atmospheric levels can result (Berthelin 1988). Soil CO2 reacts with soil water to form dissolved inorganic carbonate (DIC) through the following reaction:
Thus, changes in belowground C allocation driven by leaf-level dynamics can significantly control DIC production.
In our free air CO2 and O3 enrichment (FACE) experiment (see below) the application of a two-member mixing model to DIC chemistry can illustrate the source of increased DIC concentrations in soils. By utilizing the distinct isotopic signal of the highly depleted fumigation CO2, the measured δ13C of soil pCO2, and the measured δ13C of DIC, we can show the proportion of DIC-C that is attributable to the fumigation gas. Also, by understanding isotopic fractionation, the seasonal δ13C of DIC can be used to provide evidence for altered belowground chemistry due to changes in soil CO2 concentrations. Total system DIC, i.e. [DIC], is equal to the summed concentrations of the four inorganic carbonate species:
Though by convention, dissolved CO2 is expressed in its hydrated form as carbonic acid where:
The speciation of Eq. 2 is determined by soil solution pH. At a soil pH lower than approximately 6.4 (depending upon temperature and soil pCO2), H2CO *3 dominates DIC, with HCO −3 dominating at pH from 6.4 to 10.3. Significant isotopic fractionation occurs during the pH-mediated speciation of DIC. Soil CO2 undergoes a 1.1‰ depletion as it forms H2CO *3 , and then a 9.0‰ enrichment as it forms HCO −3 (Clark and Fritz 1997). Presumably, changes in the δ13C ratio of DIC correlate with changes in inorganic chemistry: as soil alkalinity rises, increasing the abundance of HCO −3 , the δ13C signature of DIC should become increasingly enriched relative to that of soil pCO2.
Field studies have shown that elevated atmospheric CO2 increases the concentration of soil CO2 (Andrews and Schlesinger 2001; King et al. 2001). Henry’s Law (i.e., the amount of gas that dissolves in a liquid is proportional to the partial pressure of the gas over the liquid) dictates that an increase in CO2 concentration alone will increase the equilibrium concentration of H2CO *3 by a factor of 10−1.5 (Pankow 1991). Thus, not only does elevated soil pCO2 potentially increase the rate of mineral weathering in soil, it can be expected that elevated atmospheric CO2 increases the rate of amount of mineral weathering by increasing the formation of H2CO *3 (Berner 1997; Bormann et al. 1998; Berg and Banwart 2000). H2CO *3 then readily weathers carbonates to form free Ca2+ and HCO −3 :
Through this reaction, weathering neutralizes acidic CO2 that is in the form of H2CO *3 , and keeps it in solution as HCO −3 . Silicate weathering meanwhile, extracts alkalinity from silicate minerals, as exhibited by the weathering of olivine by H2CO *3 :
and/or the hydrolysis of orthoclase feldspar by H+:
The combined processes of weathering, riverine export, and deposition of marine carbonates is a transfer and sequestration of atmospheric CO2, representing a long-term negative feedback to global warming (Drever 1994; Lackner 2002). However, the extent to which this feedback mechanism has been fully explored under experimental conditions of elevated atmospheric CO2 is limited and confounded by gaps in understanding the ecosystem-level effects of elevated atmospheric O3.
Objectives and hypothesis
The objectives of this study were to determine the effects of elevated CO2 and O3 on DIC and soil inorganic chemistry in north temperate forest ecosystems, and comment upon their role in the silicate weathering cycle in soil. This was done by quantifying DIC, tracking the δ13C signature of the ecosystem, and examining DIC speciation and δ13C fractionation. Our first hypothesis was that elevated atmospheric CO2 increases soil pCO2 and DIC concentrations, while elevated O3 results in a decrease in soil pCO2 and DIC. We further hypothesized that soil pH would decrease under elevated CO2 due to an increase in the activity of H+ and H2CO *3 .
Materials and methods
Study site
This study was conducted at the 32-ha FACTS-II Aspen FACE Project, a CO2 and O3 enrichment experiment located near Rhinelander, Wisconsin (latitude 45.6°, longitude 89.5°). The soil is a Pandus sandy loam (mixed, frigid, coarse loamy Alfic Haplorthod) derived from non-calcareous sandy to loamy glacial till. These soils were deposited over lower Proterozoic system mafic to felsic and tonalitic to granodioritic rocks. The field experiment consists of a randomized complete block design: three blocks of four treatments (control, elevated CO2, elevated O3, and elevated CO2+O3). The experimental rings are 30 m in diameter and each treatment is replicated 3 times. The rings were planted in June 1997 with trees at a 1×1 m spacing: half of the ring was planted with Populus tremuloides var. Michaux (trembling aspen) clones of differing sensitivity to CO2 and O3, a quarter was planted with a 1:1 mix of P. tremuloides and Betula papyrifera var. Marsh (paper birch), and the final quarter was planted with a mix of P. tremuloides and A. saccharum var. Marsh (sugar maple). Only the aspen and aspen-birch sections of the ring were measured in this study.
CO2 is stored and transferred to ambient-air heat exchangers that vaporize the liquefied CO2 before it is routed as a gas to individual rings. Liquefied O2 is transferred as needed to an ozone generator before it is routed as O3 to each ring. At each ring, pipes spaced uniformly around each plot release the fumigation gases in proportions dictated by real-time computer algorithms. For the 2002 growing season the CO2 fumigation period lasted from 28 May to 11 October for an exposure period of 138 days. Ambient CO2 levels averaged 360±16 μl l−1 while the concentration of elevated CO2 plots averaged 542±81 μl l−1 (J. Sober, personal communication). The O3 fumigation period lasted from 29 May to 11 October for an exposure period of 137 days. Ambient O3 levels averaged 33±14 nl l−1 while the concentration of elevated O3 plots averaged 49±24 nl l−1 (J. Sober, personal communication). A complete description of the project design, justification, hardware, and performance data can be found in Dickson et al. (2000).
Field sampling
Bi-weekly samples were collected from 1 May to 28 October 2002 using belowground collection equipment installed in 1998 and methodology as described by King et al. (2001). Soil pCO2 was withdrawn from soil gas wells at depths of 15, 30, and 125 cm in each species section and injected into serum vials sealed with butyl rubber septa. Samples were analyzed at the USDA Forestry Sciences Laboratory (Houghton, Mich.) with a Varian CP-3800 gas chromatograph equipped with a thermal conductivity detector; certified NIST-traceable gas was used to develop standard curves. Soil solution was collected using tension lysimeters installed adjacent to each soil gas well. Soil solution was withdrawn, and volume, temperature, pH, and conductivity were measured using a portable pH meter. Water samples were collected in acid-washed HDPE bottles and transported under refrigeration to the laboratory where they were acidified to pH≈2.0 with HCl, filtered (0.45 μm), and stored at 4°C. The speciation and concentration of DIC was calculated for each sampling date according to well-established carbonate equilibria reactions (Garrels and Christ 1965) using field measured soil pCO2, solution pH, and temperature. Field measured pH was corrected for degassing following the method of Suarez (1987) and used to calculate DIC.
The δ13C of the CO2 in the fumigation tank was sampled by withdrawing a 50-ml sample from the supply line with a syringe, flushing a 10-ml Exetainer vial (Labco no. 438B) with 40 ml of the gas, and then injecting the final 10 ml. The vial was then subsampled and diluted with He prior to isotopic analysis. Soil pCO2 for δ13C analysis was subsampled concurrently with soil pCO2. A set of 10-ml Exetainer vials (Labco no. 438B) that had been previously flushed with high purity He was brought into the field. Immediately before sampling, 4 ml He was withdrawn with a syringe from the vial, and 4 ml soil CO2 for δ13C analysis was injected to maintain a constant pressure. DIC for δ13C analysis was sampled from lysimeters using a modified gas evolution technique (Atekwana and Krishnamurthy 1998; Andrews and Schlesinger 2001). Using this technique, 0.25 μl of 85% phosphoric acid was added to a Labco Exetainer vial (no. 738 W) that was subsequently evacuated. Immediately upon removal of water from the tension lysimeter, 1.5 ml of the soil solution was injected into the sealed Exetainer vial with a syringe for the δ13C analysis of DIC. All samples were analyzed within the 9-day precision estimates provided by Tu et al. (2001). The δ13C analyses were performed at the Forest Ecology Stable Isotope Ratio Mass Spectrometry Laboratory in the School of Forest Resources and Environmental Science at Michigan Technological University.
C stable isotope analysis and stable C tracer
The gas isotopic analysis was performed using a ThermoFinnigan GasBench II coupled to a DELTAplus continuous flow-stable isotope ratio mass spectrometer (Bremen, Germany). IAEA-, USGS-, and NIST-certified isotopic standards were used to calibrate standard gas with a δ13C value of −49.6‰, relative to V-PDB dolomite, where δ is defined as [(ratiosample/ratiostandard)−1]×1,000. Overall analytical precision was ±0.3‰ for δ13C. The proportion of C in DIC attributable to biogenic pCO2 (fbio) was calculated using the equation: fbio=(δ1−δo)/(δ2−δo), where δ1 and δ2 are the measured δ13C values of DIC and soil pCO2, respectively, and δo the δ13C value of soil carbonate. The δ13C value of soil carbonate was assumed to be −1.7‰, halfway between the reported range of −5.3 to +1.9 as reported by Middleton et al. (1990) for southern Ontario and Taylor and Sibley (1986) for the Michigan basin, respectively. A sensitivity analysis revealed that the range of values had no impact on the results of the mixing model (results not shown). The δ13C values of H2CO *3 and HCO −3 were calculated from fractionation measurements provided by Clark and Fritz (1997). The experimental fumigation CO2 is derived from fossil fuel and had a cumulative 4-year δ13C mean (SD) of −42.1‰ (1.85) from 1999 to 2002. The treatment-level δ13C of atmospheric CO2 means (SD) in the forest canopy in 2002 were: control, −7.6‰ (0.46); +CO2, −18.5‰ (0.94); +O3, −7.2‰ (0.22); +CO2×O3, −19.0‰ (2.25). These values represent the δ13C of atmospheric CO2 after the fumigation gas was mixed with the ambient atmosphere.
Statistical analysis
Analysis required repeated measures ANOVA since the same experimental units were measured repeatedly over time. Data were analyzed with a repeated measures ANOVA model for a randomized complete block design using SAS 8.02 (Cary, N.C.) after King et al. (2001). Experimental block is a fixed effect (Dickson et al. 2000) accounting for soil differences across the site. To control for heterogeneity of variance, soil pCO2, total alkalinity, [DIC], [H2CO3*], [HCO −3 ], [CO 2−3 ], [OH−] and [H+] normal probability plots were inspected. Data were square root transformed prior to analysis, as this transformation best satisfied model assumptions. Hypothesis tests were conducted at the P≤0.05 level of confidence, and trends were reported when P<0.10. Displayed values represent the plot means averaged across the two species split plots (aspen and aspen-birch), three depths (15, 30, and 125 cm), and n=3 replicates. To simplify display, the +O3 and control treatments and the +CO2 and +CO2×O3 treatments were combined in Figs. 1 and 2. The tabular data remain unchanged.

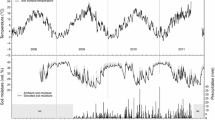
Seasonal 2002 soil temperature at 10 cm (a), soil partial pressure of CO2 (pCO2) (b), soil solution dissolved inorganic carbonate concentration ([DIC]) (c), and soil solution carbonic acid concentration ([H 2 CO * 3 ]) (d) at the FACTS II Aspen free air CO2 and O3 enrichment (FACE) experiment located near Rhinelander, Wisconsin. Ambient and elevated CO2 levels were 360±16 and 542±81 μl l−1, respectively; ambient and elevated O3 levels were 33±14 and 49±24 nl l−1, respectively. For display purposes, the +O3 and control treatments and the +CO2 and +CO2×O3 treatments were combined, respectively. Displayed values are plot means averaged across two species subplots, three depths, and n=6 replicates; bars represent SEs

Seasonal 2002 δ13C of soil pCO2 (a) and DIC (b) at the FACTS II Aspen FACE experiment. Ambient and elevated CO2 levels were 360±16 and 542±81 μl l−1, respectively; ambient and elevated O3 levels were 33 and 49±24 nl l−1, respectively. For display purposes, the +O3 and control treatments and the +CO2 and +CO2×O3 treatments have been combined, respectively. Displayed values are plot means averaged across two species subplots, three depths, and n=6 replicates; bars represent SEs. For abbreviations, see Fig. 1
Results
Soil pCO2
CO2 concentrations in the soil atmosphere were positively related to changes in soil temperature. Seasonal patterns in soil pCO2 appeared to be related to increases in mean daytime soil temperature, with peak soil pCO2 concentrations occurring with peak soil temperature in July and August (Fig. 1). This gave rise to a highly significant seasonal effect (P<0.001). The soil pCO2 level in the elevated CO2 treatments was higher on all sampling dates than in the ambient CO2 treatments, with this difference being marginal from early May until late June, and most pronounced from July to September, when the soils were warmer. Soil pCO2 concentrations in the ambient CO2 plots ranged from 7,000 μl l−1 in early May to a peak of 24,000 μl l−1 in mid July. Soil pCO2 concentrations in the elevated CO2 plots followed the same temporal pattern with a 7,500 μl l−1 baseline in early May and a mid July peak of 32,500 μl l−1. Exposure to elevated atmospheric CO2 significantly increased soil pCO2 concentrations (P=0.0239), though there were no other significant interactions. Although no other interaction was significant, there was a trend in the data toward a +O3×Time interaction (P=0.0599). This effect, however, did not appear to be distinct from the other treatments (data not shown).
Dissolved inorganic carbonate
DIC concentrations followed very similar patterns to soil pCO2 across the growing season, giving rise to a highly significant seasonal effect (P<0.001). Soil DIC concentration was greater in the elevated CO2 treatment on all sampling dates (P=0.0189), and this difference was consistent through the growing season (Fig. 1c). DIC concentrations in the ambient CO2 plots ranged from 0.33 to 1.24 μmol kg−1, while concentrations in the elevated CO2 plots ranged from 0.56 to 1.65 μmol kg−1. Depth was also significant (P<0.001), with concentrations increasing with increasing soil depth. Although there was a trend in the +CO2×O3 treatment towards higher DIC concentrations (P=0.0829) (Table 1); there were no other Species×Time or Species×Depth interactions.
To ensure that the pH correction was not affecting the calculated differences in DIC concentrations, DIC was also calculated using the uncorrected pH measurements. The results were similar to those obtained using the corrected pH (data not shown). Seasonal patterns were very similar to soil pCO2, with DIC concentrations in the elevated CO2 treatments consistently higher than those in the ambient treatments (P=0.1141). However, the use of uncorrected pH measurements in the DIC calculation overstated DIC concentrations by 6–24% compared to the use of corrected pH measurements.
Inorganic carbonate chemistry
The concentrations of H2CO3*, HCO −3 , CO 2−3 , OH− and total alkalinity (AT) followed seasonal patterns similar to that of soil pCO2 (Fig. 1d, results only shown for H2CO3*). Exposure to elevated CO2 consistently increased ion concentrations and AT (Table 1). The opposite effect, however, was observed for [H+], where exposure to elevated CO2 reduced the concentration. Though these changes were significant for [H2CO *3 ] (P=0.0281), they were not statistically significant for any other ionic species. All ionic species other than [H2CO *3 ] exhibited a highly significant depth effect (Table 2): the concentration of H2CO *3 and H+ increased with soil depth, while the concentration of HCO −3 , CO 2−3 , OH− and AT decreased with soil depth. Time was also highly significant for AT and all ionic species, except [H+] and [OH−], with peak concentrations occurring in August. These results were driven by changes in soil pCO2 with depth, time of year, and treatment.
δ13C of soil pCO2
The heavily depleted δ13C signal of the fumigation gas led to significant (P<0.001) differences in the isotopic ratio of soil pCO2 between the ambient and elevated CO2 treatments (Table 3). The average δ13C of soil pCO2 values in ambient CO2 plots was measured at −21.5‰ in May, and became steadily more depleted to −23.5‰ in early July (Fig. 2a). Values then became steadily enriched into October. The δ13C of soil pCO2 followed a different pattern in the elevated CO2 treatment. δ13C values were measured at −30.0‰ in May, and became consistently depleted to values of −34.9‰ in late July. Values then became slightly enriched into October. Though values in the elevated atmospheric CO2 treatments were already depleted by 9.0‰, they became increasingly depleted relative to ambient CO2 treatments as the growing season progressed. These patterns gave rise to a significant seasonal time effect (P<0.001) and +CO2×Time effect (P<0.001). A strong species effect was observed, with the δ13C of soil pCO2 becoming more depleted in aspen-birch subplots than in pure aspen subplots (P=0.0058) (data not shown). A strong depth effect was also observed, with δ13C values becoming more depleted with depth in the soil profile (P=0.0086) (data not shown). The δ13C of the fumigation gas was consistent throughout the growing season and did not explain the isotopic variation seen in the CO2 fumigated plots.
δ13C of DIC
The depleted δ13C signal in the fumigation gas also led to significant differences in isotopic ratios between ambient and elevated CO2 plots for DIC (P<0.001), though no differences were seen for elevated O3. δ13C of DIC in ambient CO2 plots was measured at −22.0‰ in May, and became consistently enriched to values of −19.7‰ in October (Fig. 2b). δ13C values followed a different pattern in the elevated CO2 treatment, which also could not be explained by the seasonal δ13C of the fumigation gas. δ13C values were measured at −29.0‰ in early May, and became enriched to a value of −27.6‰ in late May (Fig. 2b). Values then became consistently depleted to −30.0‰ in October. This gave rise to a highly significant time effect (P<0.001). While δ13C values in elevated CO2 treatments became more depleted relative to those in the ambient CO2 plots, there were no significant time interactions to explain this tendency. No Species, Species×Time, Depth, or Species×Depth interactions were significant. As δ13C of H2CO3 and HCO −3 were calculated from δ13C of soil pCO2, these trends follow those of soil pCO2 and thus P-values are not displayed, although annual means are displayed in Table 3.
Discussion
Elevated atmospheric CO2 has been hypothesized to increase soil pCO2 through increased fine root production and turnover, and resulting increases in the respiration of autotrophs (RA) and heterotrophs (RH). Fine root production (measured in 1999) in the same stands of P. tremuloides increased 96% relative to the control under experimental atmospheric CO2 enrichment (King et al. 2001). P. tremuloides fine root biomass was also shown to increase by 52% under elevated atmospheric CO2 in an open top chamber study (Pregitzer et al. 2000). An increase in fine root biomass of 86% has also been reported for a similar forested FACE experiment of Pinus taeda (Matamala and Schlesinger 2000). While these increases in fine root standing stock represent increased levels of RA, they also facilitate increases in RH. For the 1999 data, King et al. (2001) reported a 139% increase in dead-root stock under elevated CO2, and the authors have concluded that elevated atmospheric CO2 increases fine root turnover (Kubiske et al. 1998; Pregitzer et al. 1995, 2000). As most microbial communities are C limited (Zak and Pregitzer 1998), this enhanced addition of organic matter through organic acids, root turnover, and increased microbial metabolism likely increased RH as well (Zak et al. 2000).
Soil pCO2 concentrations, averaged across soil depths and species subplots increased by 14% under elevated CO2. Similar increases under elevated CO2 have previously been reported (Andrews and Schlesinger 2001; King et al. 2001). With increasing concentrations of soil CO2, Eq. 1 is driven to the right, increasing the production of DIC. The increase in DIC was calculated to be 22% under elevated CO2. Though DIC concentrations were calculated with pH values that were corrected for tension lysimeter degassing, it is noted that DIC calculations utilizing uncorrected pH measurements may overstate DIC concentrations by up to 25%, since the concentration of DIC increases with pH (Fig. 3). The concentration of H2CO *3 increased by 22% due to increased concentrations of soil CO2, as governed by the Henry’s law constant, KH. However, while the concentration of H2CO *3 increased under elevated CO2, it made up a proportionately smaller percentage of DIC–C. The percentage of DIC–C existing as HCO −3 increased from 11% under ambient CO2 to 17% under elevated CO2. This proportional increase in HCO −3 under elevated CO2 is accompanied by increases in the concentrations of carbonate (CO 2−3 ), hydroxide (OH−), and total alkalinity, as well as a decrease in the concentration of H+.

Open system equilibrium diagram for ambient CO2 FACE conditions of 16.3°C, pH of 5.16 and soil pCO2 of 10−1.87 (13,500 μl l−1). Ambient and elevated CO2 levels were 360±16 and 542±81 μl l−1, respectively; ambient and elevated O3 levels were 33±14 and 49±24 nl l−1, respectively. Inset illustrates equilibrium model shifts under the elevated CO2 FACE system conditions of 16.3°C, pH of 5.26, and soil pCO2 of 10−1.84 (14,500 μl l−1). For abbreviations, see Fig. 1
Taken together these results support our first hypothesis that elevated atmospheric CO2 can be shown to increase soil pCO2 and concentrations of DIC. Trends in the data also suggest support for our hypothesis that elevated O3 reduces soil pCO2 and DIC. The effects of elevated O3, however, are not statistically distinguishable from the other treatments. These data do not support our second hypothesis, that soil acidity will increase under elevated CO2 due to increased concentrations of H+ and H2CO *3 . The data instead show the opposite effect, that elevated atmospheric CO2 increases soil alkalinity, and that soil pH is buffered by changes in soil inorganic carbonate chemistry under elevated CO2. These observations are explained by an open system model of equilibrium carbonate chemistry (Fig. 3).
In the ambient CO2 model, the open system equilibrium model is driven by the external soil pCO2 concentration of 13,500 μl l−1. Through carbonate equilibria equations and Henry’s Law, the concentration of external soil pCO2 then dictates [H2CO *3 ], which remains constant across the pH range. While [H2CO *3 ] remains constant, total DIC increases after pH 5.0 because of the increasing contribution of HCO −3 until the species contribute an equal portion of C to DIC. Under elevated atmospheric CO2, the system is driven by the external soil pCO2 concentration of 14,500 μl l−1. This greater concentration of external soil pCO2 under elevated CO2 then establishes a greater concentration of H2CO *3 . Figure 3 illustrates the result of these shifts in the open system equilibrium model. Under elevated soil pCO2, the concentration of H2CO *3 increases, as does the concentration of HCO −3 . However, due to a small increase in pH the elevated CO2 system equilibrium point moves to the right as well, explaining the greater proportion of HCO −3 -derived C in DIC (Fig. 3).
Seasonal δ13C data also support the assumptions of the open system equilibrium model. Distribution models show that system pH governs the distribution of carbonate species in water (Clark and Fritz 1997). In the FACTS II Aspen FACE experiment, H2CO *3 is the dominant C-bearing species, with the importance of HCO −3 increasing with pH. Fractionation models show that this pH-mediated speciation results in a temperature-dependent fractionation, with δ13C values becoming depleted by approximately 1.1‰ from CO2(g) to H2CO *3 , and enriched 9.0‰ from H2CO *3 to HCO −3 (Zhang et al. 1994; Clark and Fritz 1997). Thus, with a δ13C of soil pCO2 of −21‰, DIC having a pure H2CO *3 or HCO −3 source would be −22.2 and −13.2‰, respectively. Under ambient CO2, the δ13C of DIC is very similar to soil pCO2, suggesting very little influence of HCO −3 on δ13C. However, under elevated CO2, the measured δ13C of DIC is enriched compared to soil pCO2, suggesting a larger contribution of the enriched HCO −3 to total DIC. It is unlikely that this pattern was complicated by contamination from local carbonate minerals with δ13C ranges from −5.3 to 1.9‰ as reported by Middleton et al. (1990) for southern Ontario and Taylor and Sibley (1986) for the Michigan basin. Carbonates have already been weathered from the upper meter of this soil (D. R. Zak, personal communication), and laboratory tests for the presence of inorganic carbonate were negative (data not shown). Furthermore, seasonal δ13C of soil pCO2 and DIC showed no influence of enrichment that could be caused by rising water tables or hydraulic lift carrying carbonate-contaminated water from deeper soil to the near surface. Measured δ13C of DIC and soil pCO2 in the elevated CO2 treatments agrees well with values calculated in the manner of Stumm and Morgan (1981) as displayed in Amiotte-Suchet et al. (1999) and values from another FACE system (Andrews and Schlesinger 2001). Values in the ambient CO2 treatments also fall within the range of natural variation found in forest ecosystems (Kendall and McDonnel 1998).
Taken together, these data suggest to us that elevated atmospheric CO2 alters ecosystem inorganic carbonate chemistry. A 50% increase in atmospheric CO2 resulted in a 14% increase in soil pCO2 concentrations. In the absence of carbonate, soil pCO2 would account for all C present in DIC (Kendall and McDonnel 1998). With the use of a mixing model and assumed values of soil carbonate (see Materials and methods), 95–100% of DIC–C is attributable to biogenic pCO2, establishing elevated atmospheric CO2 as the direct cause of increased DIC concentration and altered speciation (Table 3). This finding also provides evidence that short-term terrestrial C cycling may be linked to the long-term silicate C weathering cycle through a time step shorter than may have been previously assumed. Soil pCO2 increased by 14% under elevated CO2 and [H2CO *3 ] increased by 14% also, as expected by KH. However, calculated DIC concentrations increased by 22%, with a 78% increase in [HCO −3 ], a 294% increase in [CO 2−3 ], and a 210% increase in AT. The increases in [DIC] have likely contributed to the calculated increase in speciation and AT. These increases may also be due in part to enhanced primary mineral weathering by increased production of H2CO *3 . It is possible that weathering of silicates (Eqs. 4, 5) increases alkalinity through the production of HCO −3 and OH−. Sufficient amounts of P- or S-bearing minerals could result in the weathering of sulfate and phosphate in concentrations high enough to neutralize H+, explaining the pattern seen in [H+] under elevated CO2 in Table 3. It is even possible that carbonate minerals such as calcite (Eq. 3) or dolomite could contribute excess inorganic carbonate, though this is unlikely without the presence of soil inorganic carbonate.
Conclusions
This study has shown that elevated atmospheric CO2 increased soil pCO2, [DIC], and [H2CO *3 ]: the effects of elevated O3 were transient or muted, and require further study. In addition to increasing concentrations of C in soil solution, the study has shown that elevated atmospheric CO2 alters system inorganic carbonate chemistry by increasing alkalinity, with the potential to increase weathering rates of primary minerals. Given an estimated annual oceanic DIC input of 0.4×1015 g C year−1 (Schlesinger 1997), and extrapolating a 22% increase in soil solution DIC concentration as reported here to be consistent across soil types throughout the world, we calculate a potential 0.1×1015 g C year−1 increase in global DIC delivery to the ocean, a rate consistent with estimates cited in Houghton (2003). Though DIC is a minor global CO2 flux compared to net primary production and respiration (60×1015 g C year−1, Schimel 1995), it represents a potential long-lived sequestration reservoir in deep ocean sediments. This extrapolation is not intended to be a robust parameter for global C modeling: however, it illustrates that aggrading forest ecosystems may be used to capture and sequester atmospheric CO2 through inorganic processes.
References
Amiotte-Suchet P, Aubert D, Probst JL, Gauthier-Lafaye F, Probst A, Andreux F, Viville D (1999) δ13C pattern of dissolved inorganic carbon in a small granitic catchment: the (2000) Strengbach case study (Vosges mountains, France). Chem Geol 159:129–145
Andrews JA, Schlesinger WH (2001) Soil CO2 dynamics, acidification, and chemical weathering in a temperate forest with experimental CO2 enrichment. Glob Biogeochem Cycles 15(1):149–162
Atekwana EA, Krishnamurthy RV (1998) Seasonal variations of dissolved inorganic carbon and d13C of surface waters: application of a modified gas evolution technique. J Hydrol 205:265–278
Berg A, Banwart SA (2000) Carbon dioxide mediated dissolution of Ca-feldspar: implications for silicate weathering. Chem Geol 163:25–42
Berner RA (1997) The rise of plants and their effect on weathering and atmospheric CO2. Science 276:544–546
Berthelin J (1988) Microbial weathering processes in natural environments. In: Lerman A, Meybeck M (eds) Physical and chemical weathering in geochemical cycles. Kluwer, Dordrecht, pp 33–59
Bormann BT, Wang D, Bormann FH, Benoit G, April R, Snyder R (1998) Rapid, plant-induced weathering in an aggrading experimental ecosystem. Biogeochemistry 43:129–155
Chameides WL, Kasibhatla PS, Yienger J, Levy H II (1994) Growth of continental-scale, metro-agro-plexes, regional ozone pollution, and world food production. Science 264:74–77
Chappelka AH, Samuelson LJ (1998) Ambient ozone effects on forest trees in the Eastern United States: a review. New Phytol 139:91–108
Clark ID, Fritz P (1997) Environmental isotopes in hydrogeology. CRC, New York
Coleman MD, Isebrands JG, Dickson RE, Karnosky DF (1995) Photosynthetic productivity of aspen clones varying in sensitivity to tropospheric ozone. Tree Physiol 15:585–592
DeLucia EH, Hamilton JG, Naidu SL, Thomas RB, Andrews JA, Finzi A, Lavine M, Matamala R, Mohan JE, Hendrey GR, Schlesinger WH (1999) Net primary production of a forest ecosystem with experimental CO2 enrichment. Science 289:1177–1179
Dickson RE, Lewin KF, Isebrands JG, Coleman MD, Heilman WE, Riemenschneider DE, Sober J, Host GE, Zak DR, Hendrey GR, Pregitzer KS, Karnosky DF (2000) Forest atmosphere carbon transfer and storage (FACTS-II). The aspen free-air CO2 and O3 enrichment (FACE) project: an overview. USDA Forest Service. General Technical Report NC—214
Drever JI (1994) The effect of land plants on weathering rates of silicate minerals. Geochim Cosmochim Acta 58(10):2325–2332
Fowler D, Cape JN, Coyle M, Flechard C, Kuylenstierna J, Hicks K, Derwent D, Johnson C, Stevenson D (1999) The global exposure of forests to air pollutants. Water Air Soil Pollut 116:5–32
Garrells RM, Christ CL (1965) Solutions, minerals, and equilibria. Harper-Collins, New York
Houghton RA (2003) Why are estimates of the terrestrial carbon balance so different? Global Change Biol 9:500–509
Houghton JT, Meira Filho LG, Callander BA, Harris N, Kattenberg A, Maskell K (1996) Climate change 1995: the science of climate change Cambridge University Press, Cambridge
Karnosky DF, Gagnon ZE, Dickson RE, Coleman MD, Lee EH, Isebrands JG (1996) Changes in leaf growth, leaf abscission, and biomass associated with seasonal tropospheric ozone exposures of Populus tremuloides clones and seedlings. Can J For Res 26:23–37
Kendall C, McDonnel JJ (1998) Isotope tracers in catchment hydrology. Elsevier, New York
King JS, Pregitzer KS, Zak DR, Sober J, Isebrands JG, Dickson RE, Hendrey GR, Karnsoky DF (2001) Fine-root biomass and fluxes of carbon in young stands of paper birch and trembling aspen as affected by elevated atmospheric CO2 and tropospheric O3. Oecologia 128:237–250
Kubiske ME, Pregitzer KS, Zak DR, Mikan CJ (1998) Growth and C allocation of Populus tremuloides genotypes in response to atmospheric CO2 and soil N availability. New Phytol 140:251–260
Lackner KS (2002) Carbonate chemistry for sequestering fossil carbon. Annu Rev Energy Environ 27:193–232
Landry LG, Pell EJ (1993) Modification of Rubisco and altered proteolytic activity in O3-stressed hybrid poplar (Populus maximowizii × trichocarpa). Plant Physiol 101:1355–1362
Loya WM, Pregitzer KS, Karberg NJ, King JS, Giardina CP (2003) Reduction of soil carbon formation by tropospheric ozone under increased carbon dioxide levels. Nature 425:705–707
Marenco A, Gouget H, Nedelec P, Pages JP, Karcher F (1994) Evidence of a long-term increase in tropospheric ozone from Pic du Midi series: consequences: positive radiative forcing. J Geophys Res 99(D8):16617–16632
Matamala R, Schlesinger WH (2000) Effects of elevated atmospheric CO2 on fine root production and activity in a temperate forest ecosystem. Global Change Biol 6:967–980
Middleton K, Coniglio M, Frape SK (1990) Burial dedolomitization, Middle Ordivician carbonates, southwestern Ontario. AAPG Bull 74:1308
Pankow JF (1991) Aquatic chemistry concepts. Lewis, Chelsea
Pell EJ (1987) Ozone toxicity-is there more than one mechanism of action? In: Hutchinson TC, Meema KM (eds) Effects of atmospheric pollutants on forests, wetlands, and agricultural ecosystems. (NATO ASI series, vol G16) Springer, Berlin Heidelberg New York, pp 229–240
Phillips RL, Zak DR, Holmes WE, White DC (2002) Microbial community composition and function beneath temperate trees exposed to elevated atmospheric carbon dioxide and ozone. Oecologia 131:236–244
Pregitzer KS, Zak DR, Curtis PS, Kubiske ME, Teeri JA, Vogel CS (1995) Atmospheric CO2, soil nitrogen, and turnover of fine roots. New Phytol 129:579–585
Pregitzer KS, Zak DR, Masiaz J, DeForest J, Curtis PS, Lussenhop J (2000) Interactive effects of atmospheric CO2 and soil-N availability on the fine roots of Populus tremuloides. Ecol Appl 10:18–33
Reich PB, Ellsworth DS, Kloeppel BD, Fownes JH, Gower ST (1990) Vertical variation in canopy structure and CO2 exchange of oak-maple forests influence of ozone, nitrogen, and other factors on simulated canopy carbon gain. Tree Physiol 7:329–345
Sage RF (1994) Acclimation of photosynthesis to increasing atmospheric CO2: the gas exchange perspective. Photosynth Res 39:351–368
Schimel DS (1995) Terrestional ecosystems and the carbon cycle. Global Change Biol 1:77–91
Schlesinger WH (1997) Biogeochemistry: an analysis of global change. Academic Press, New York
Stumm W, Morgan JJ (1981) Aquatic chemistry. Wiley, New York
Suarez DL (1987) Prediction of pH errors in soil-water extractors due to degassing. Soil Sci Soc Am J 51:64–67
Taylor TR, Sibley DF (1986) Ferroan dolomite in the Trenton formation, Ordovician Michigan Basin. Sedimentology 33:61–86
Tolbert NE, Zelitch I (1983) Carbon metabolism. In: Lemon ER (eds) CO2 in plants. Westview, Boulder, Colo., pp 71–88
Tu KP, Brooks PD, Dawson TE (2001) Using septum-capped vials with continuous flow isotope ratio mass spectrometric analysis of atmospheric CO2 for Keeling plot applications. Rapid Commun Mass Spectrom 15
Zak DR, Pregitzer KS (1998) Integration of ecophysiological and biogeochemical approaches to ecosystem dynamics. In: Pace M, Groffman P (eds) Successes, limitations, and frontiers in ecosystem science. Springer, Berlin Heidelberg New York, pp 372–403
Zak DR, Pregitzer KS, King JS, Holmes WE (2000) Elevated atmospheric CO2, fine roots and the response of sol microorganisms: a review and hypothesis. New Phytol 147:201–222
Zhang J, Quay PD, Walbur DO (1994) Carbon isotope fractionation during gas-water exchange and dissolution of CO2. Geochim Cosmochim Acta 59:107–114
Acknowledgements
This study was aided by the help of numerous people: J. Taylor, E. Nauertz, R. Ketterer, W. Loya, M. Porter, R. Peryam, K. Larsen, J. Ashby, J. Eikenberry, and A. Johansen. This research was supported by the US Department of Energy’s Office of Science ( Program for Ecosystem Research and National Institute for Global Environmental Change), the USDA Forest Service (Northern Global Change Program and North Central Research Station), the National Science Foundation (DEB, DBI/MRI), and the USDA National Research Initiatives Competitive Grants Program. G. Hendrey, K. Lewin, J. Nagey, D. Karnsoky, and J. Sober have been instrumental in the successful implementation of this long-term study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Karberg, N.J., Pregitzer, K.S., King, J.S. et al. Soil carbon dioxide partial pressure and dissolved inorganic carbonate chemistry under elevated carbon dioxide and ozone. Oecologia 142, 296–306 (2005). https://doi.org/10.1007/s00442-004-1665-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00442-004-1665-5