
Abstract
Salivary gland function is severely disrupted by radiation therapy used to treat patients diagnosed with head and neck cancer and by Sjögren’s syndrome. The resulting condition, which results in xerostomia or dry mouth, is due to irreversible loss of the secretory acinar cells within the major salivary glands. There are presently no treatments for the resolution of xerostomia. Cell-based approaches could be employed to repopulate acinar cells in the salivary gland but investigations into potential therapeutic strategies are limited by the challenges of maintaining and expanding acinar cells in vitro. We investigate the encapsulation of salivary gland cell aggregates within PEG hydrogels as a means of culturing secretory acinar cells. Lineage tracing was used to monitor the fate of acinar cells isolated from murine submandibular gland (SMG). Upon initial formation in vitro, SMG aggregates comprise both acinar and duct cells, with the majority cells of acinar origin. With longer culture times, acinar cells significantly decreased the expression of specific markers and activated the expression of keratins normally found in duct cells. A similar acinar-to-duct cell transition was also observed in vivo, following duct ligation injury. These results indicate that under conditions of stress (mechanical and enzymatic isolation from glands) or injury (duct ligation), salivary gland acinar cells exhibit plasticity to adopt a duct cell phenotype.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Salivary gland function is critical for oral health, and disruptions in saliva secretion have adverse secondary effects. Chronic salivary gland dysfunction is caused by radiation therapy used to treat head and neck cancers, total body irradiation for allogeneic stem cell transplantation and by systemic autoimmune diseases, such as Sjögren’s syndrome. There is presently no permanent treatment for restoring saliva secretion in these patients.
Differentiated salivary gland acinar cells, like pancreatic acinar cells and liver hepatocytes, are primarily maintained in the gland through self-duplication (Aure et al. 2015a, b; Desai et al. 2007; Emmerson et al. 2018). A major limitation for the design of cell-based strategies to regenerate the salivary glands is the difficulty of maintaining and expanding acinar cells in vitro. Recent evidence that acinar-like cells can be derived from human minor salivary gland biopsies (Jang et al. 2015) indicates that in vitro limitations may be overcome through modification of culture conditions. We previously investigated whether enzymatically degradable poly(ethylene glycol) (PEG)-based hydrogels could support in vitro culture of primary acinar cells. SMG cells encapsulated as aggregates into hydrogels exhibited lumen formation and cellular organization of apical and basolateral tight junction markers, as well as expression of the basement membrane proteins laminin and collagen IV (Shubin et al. 2017). However, expression of the critical acinar cell transcription factor Bhlha15 (Mist1), was significantly reduced.
In response to tissue injury, or in vitro primary cell culture, pancreatic acinar cells lose their secretory phenotype and activate expression of duct cell markers, a process known as acinar-to-ductal metaplasia (Jensen et al. 2005; Means et al. 2005; Pan et al. 2013). To investigate whether acinar-to-ductal metaplasia explains the loss of the acinar cell phenotype in encapsulated SMG aggregates, this study used a genetic labeling system based on the Mist1CreERT2;Rosa26tdTomato mouse strain to investigate the fate of acinar cells in vitro and in vivo in response to environmental stress induced by duct ligation.
Materials and methods
Four-arm poly(ethylene glycol) (PEG)-norbornene synthesis
Four-arm 20 kDa PEG-OH (Jenkem Technologies) was functionalized with norbornene (PEG-OH-Norb) using N,N′-dicyclohexylcarbodiimide (DCC) coupling as previously described (Fairbanks et al. 2009; Shubin et al. 2015).
Peptide synthesis
The peptide GKKCGPQGIWGQCKKG (MMP degradable peptide) was synthesized and characterized as previously described (Shubin et al. 2017). Peptide purity was analyzed via 205 nm absorbance measured in ddH2O with an Evolution UV/Vis detector (Thermo Scientific) and compared with predicted values (Anthis and Clore 2013). Thiol-functionalization was determined using Ellman’s reagent (Alexis Biochemical). Absorbance was analyzed at 405 nm with a Tecan Infinite M200 plate reader and compared with a cysteine (Alfa Aesar) standard curve.
Mice
Mist1CreERT2 mice (Pin et al. 2001) were crossed with the Rosa26tdTomato (Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J) strain (obtained from Jackson Laboratory) and maintained on a C57BL/6L background. Standard genotyping was performed. Mice were maintained on a 12-h light/dark cycle. Food and water were provided ad libitum. All procedures and protocols were conducted in accordance with the US Public Health Service’s Policy on Humane Care and Use of Laboratory Animals and were approved by the University Committee on Animal Resources at the University of Rochester Medical Center.
Duct ligation
Mist1CreERT2;R26tdTomato male mice (6–8 weeks, n = 3) were given one dose of tamoxifen (0.25 mg/g body weight) (MP Biomedicals) by gavage 3 days before duct ligation. On the day of surgery, mice were pre-emptively given buprenorphine SR (1 mg/kg, Wildlife Pharmaceuticals) subcutaneously as analgesia and then anesthetized with an intraperitoneal injection of ketamine HCl (80 mg/kg, JPH Pharmaceuticals) and xylazine (8 mg/kg, Lloyd Laboratories) in saline. A ventral incision was made on the left side of the neck to isolate the main secretory duct of the left SMG. The SMG main duct was ligated using a titanium hemostatic clip (#R9180, Vitalitec Int.). The right contralateral gland served as the sham-operated control. After 14 days, mice were re-anesthetized and the clip was removed. Following the de-ligation, the SMGs were either immediately isolated or allowed to regenerate for 14 days. Tissue was isolated and fixed in 4% paraformaldehyde at 4 °C overnight. The tissue was processed using a Tissue-Tek VIPTM processing machine (Sakura Finetek USA, Inc.) before paraffin embedding. Sections (5 μm) were cut using a Leica RM2125 rotary microtome and collected on Superfrost Plus slides (Thermo Fisher Scientific).
SMG cell isolation and aggregate formation
Primary SMG cells were isolated from Mist1CreERT2;R26tdTomato male or female mice as described (Rugel-Stahl et al. 2012; Shubin et al. 2015). Briefly, SMG were surgically removed, finely minced and dissociated in 5-mL Hank’s balanced salt solution (GIBCO) with CaCl2 and MgCl2, 1 mg/mL collagenase type II (GIBCO) and 5 μg/mL hyaluronidase (Sigma) at 37 °C for 40 min with shaking. Cells were washed in 5-mL Dulbecco’s phosphate buffered saline (DPBS, GIBCO) and incubated in 0.05% trypsin (GIBCO) for 4 min at room temperature. Cells were passed through a 40-μm mesh filter and resuspended in 5 mL complete media DMEM:F12 (1:1, GIBCO) with Glutamax (1x GIBCO), antibiotic-antimycotic solution (100 I.U./mL penicillin, 100 μg/mL streptomycin, 0.25 μg/mL amphotericin B, GIBCO), N2 supplement (1x Invitrogen), 10 μg/mL insulin (Life Technologies), 1 μM dexamethasone (Sigma), 20 ng/mL epidermal growth factor (EGF, Life Technologies) and 20 ng/mL basic fibroblast growth factor (bFGF, Life Technologies). Cells were seeded at a density of approximately 1.2 × 106 cells/mL in 60-mm non-tissue culture-treated petri dishes (5 ml total) and incubated in 37 °C, 5% CO2 for 48 h to allow aggregate formation. For analysis of gene expression, aggregates were seeded into a 24-well non-tissue culture-treated plate in 1 mL of media per well at a concentration of 1.5 × 105 cells/mL.
SMG cell aggregate encapsulation
Primary SMG cells or SMG cell aggregates were encapsulated within hydrogels using thiol-ene photopolymerization. The pre-polymerization solution consisted of 2-mM norbornene-functionalized 4-arm PEG-hydroxyl macromer and 4 mM of MMP-degradable peptide, 0.028 wt% of the photo-initiator lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP), synthesized as described (Fairbanks et al. 2009) and 0.1 mg/mL laminin in DPBS. The total concentration of functional groups in the pre-polymerization solution was 8-mM norbornene and 8-mM thiol. SMG cell aggregates or primary gland isolates were resuspended in the pre-polymerization solution at a concentration of ~ 5 × 106 cells/mL and pipetted in 30 μL of aliquots into 1-mL syringes with the tips cutoff. Due to rapid sedimentation of the aggregates, each syringe was immediately polymerized using 365-nm light (power ~ 5 mW/cm2). After 3 min of UV exposure, each polymerized hydrogel was placed in 1 mL of aggregate media in a 24-well plate. Media was exchanged every 2 days and fresh EGF and bFGF were added to media stocks every 4 days.
RNA extraction, reverse transcription and qPCR
Primary SMG isolates (day 0) or SMG aggregates (day 2) were pelleted by centrifugation and lysed in 350-μL TRK lysis buffer (Omega Biotek) with β-mercaptoethanol (β-ME, 20 μL per 1 mL lysis buffer). Samples were transferred to a homogenizer column (Omega Biotek) and centrifuged at 10,000 RCF for 2 min. Sample flow through was precipitated with an equivalent volume of 70% ethanol (~ 350 μL) and transferred to an E.Z.N.A. RNA purification column (Omega Biotek). RNA was purified and isolated according to manufacturer’s instructions. RNA concentration and quality were determined using a NanoVue UV-vis spectrophotometer. Samples were reverse transcribed into cDNA using the iScript™ cDNA synthesis kit (BioRad). QPCR was performed on a CFX96 real-time system (BioRad) using SsoFast Evogreen Supermix (BioRad) according to the manufacturer’s instructions, with 10 μM of forward and reverse primers (Supplemental Table 1) and cDNA. Gene expression was analyzed using the Pfaffl method with LE32 as the housekeeping gene (Pfaffl 2001).
Fixation, embedding and immunofluorescence staining
Hydrogel fixation, embedding and sectioning
At specified time points, hydrogels were fixed in 4% PFA for 45 min at room temperature with shaking, then washed with 1-mL DPBS 3× for 5 min and incubated overnight at 4 °C in 1 mL of 1% poly(vinyl alcohol) (PVA, MW: 146,000-168,000, Sigma-Aldrich) to improve cryosectioning (Ruan et al. 2013). Hydrogels were embedded in optimal cutting temperature solution (OCT, Tissue-Tek). Ten-micrometer sections were cut using a HM 550 cryostat (Micron) and mounted. Slides were dried overnight at 37 °C and stored at − 20 °C prior to use.
Submandibular gland fixation, embedding and sectioning
Mist1CreERT2;R26tdTomato mice were administered tamoxifen (0.25 mg/g body weight) by gavage daily for 4 days (Aure et al. 2015b). Mice (n = 3) were sacrificed for gland removal 3 days after the last tamoxifen gavage. Mist1CreERT2;R26tdTomato mice not treated with tamoxifen served as controls to detect non-specific recombination. SMG were fixed overnight in 4% PFA. For frozen sections, whole glands were transferred through a sucrose gradient (5%, 10%, 15% sucrose for 30 min each) and incubated in 30% sucrose: OCT (50:50) solution overnight. Samples were embedded in OCT and sectioned at 10 μm using a cryostat. For paraffin sections, SMG was processed in a Tissue-Tek VIPTM processing machine (Sakura Finetek USA, Inc.) and embedded in paraffin. Sections (5 μm) were cut using a Leica RM2125 rotary microtome.
Immunofluorescence staining
Frozen sections were blocked in 5 vol% normal donkey serum (Jackson Immunoresearch), 1 wt% bovine serum albumin (Sigma) and 0.1 vol% Triton X-100 (Sigma) for 1 h at room temperature and samples were washed in PBS for 5 min. Sections were incubated with primary antibodies diluted in 1 wt% BSA at 4 °C overnight. Primary antibodies are aquaporin 5 (AQP5) (Santa Cruz), keratin 5 (K5) (Biolegend), keratin 7 (K7) (Abcam), keratin 19 (K19) (Santa Cruz), NKCC1 (Santa Cruz), pan-keratin (Dako) and red fluorescent protein (RFP) (Rockland) (Supplemental Table 2). After overnight incubation, sections were washed in PBS (3 × 5 min) and incubated with corresponding secondary antibodies (Supplemental Table 2) in 1 wt% or 0.1 wt% BSA for 1 h at room temperature.
Paraffin sections were deparaffinized and rehydrated. Sections were heated in antigen retrieval buffer (10 mM Tris base, 1 mM EDTA) for 10 min. After cooling, sections were blocked with 10 vol% normal donkey serum (Jackson Immunoresearch) in PBS containing 0.1 wt% bovine serum albumin (Sigma), before incubating in primary antibody overnight at 4 °C. Primary antibodies are K7 (Abcam), K19 (Santa Cruz) and RFP (Rockland). The following day, sections were rinsed in PBS (3 × 5 min) and incubated with corresponding secondary antibodies (Online Resource Table 2). Nuclei were stained with DAPI (Thermo Fisher Scientific). Sections were mounted in Shandon Immu-Mount (Thermo Fisher Scientific).
5-Ethynyl-2′-deoxyuridine (EdU) staining
Hydrogel-encapsulated SMG aggregates were treated with 10-μM EdU for 3 h prior to fixation with 4 wt% PFA for 45 min, washed twice with 3 wt% BSA in DPBS and permeabilized with 0.5 vol% of Triton X-100 for 30 min. Samples were washed 2× with 3 wt% BSA in DPBS and labeled with the Click-iT® Plus EdU AlexaFluor® 647 imaging kit (ThermoFisher) and Hoechst 33342 as a nuclear counterstain. Hydrogel samples were imaged using a FV1000 laser scanning confocal microscope (Olympus) with a 40× objective. Each hydrogel was imaged in 3 separate 45-μm z-stacks consisting of 19 images each separated by 2.5 μm.
Imaging
Whole hydrogel samples were placed in glass-bottom microwell dishes (Maktech) and 45-μm z-stacks consisting of individual images every 2.5 μm were taken using a 40× oil immersion objective lens, or 10-μm z-stacks with slices every 1 μm, using an 100× objective. For slides, single slices or 15-μm z-stacks with 2.5-μm spacing between slice were taken using a 40× or oil immersion objective lens. All stains were performed on sections from at least 3 separate hydrogels or SMGs with 3 images per sample. For tissue imaging, single slices were taken with 40× objective and 3× zoom on an Olympus FV1000. Negative controls were imaged to ensure low non-specific background staining.
Image analysis
The number of total cells and tdTomato+ cells was quantified in z-stacks using VisioPharm software. Briefly, the total area of DAPI and tdTomato signals was calculated using Bayesian classification. The total area of DAPI and tdTomato fluorescence per slice was divided by average nuclear and cell size to estimate the number of total cells and tdTomato+ cells, respectively. The percentage of acinar cells was determined by dividing the number of tdTomato+ cells by the total number of cells. Co-localization of tdTomato with AQP5, NKCC1, K7, or K19 was quantified by manual counting using images from 2 to 3 mice or aggregate encapsulations.
Statistical analysis
Statistical analyses were performed via 2-way ANOVA with Sidak’s multiple comparisons test with 95% confidence interval, one-way ANOVA with Tukey’s post-hoc analysis for multiple comparisons, or unpaired T test with Welch’s correction using Prism software (Graphpad) with p < 0.05 as the threshold for statistical significance. All graphs are mean ± standard deviation.
Results
Salivary gland aggregates include acinar cells
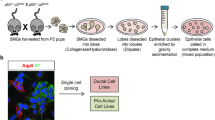
We previously used the inducible Mist1CreERT2;R26tdTomato mouse strain to label and track differentiated salivary gland acinar cells and their descendants (Aure et al. 2015b). To evaluate the fate of acinar cells in vitro, the tdTomato reporter allele was activated by inducing Mist1CreERT2;R26tdTomato mice with tamoxifen prior to SMG cell isolation. Dissociated, primary SMG cells were cultured under non-adherent conditions for 2 days to allow aggregates to form. Both tdTomato+ acinar cells and non-labeled duct cells are present in day 2 aggregates (Fig. 1 and Online Resource S1). Aggregates were encapsulated in PEG hydrogels after 2 days of culture.

Aggregate formation and PEG hydrogel encapsulation. a Paraffin section of SMG from Mist1CreERT2;R26tdTomato mouse following tamoxifen administration to activate expression of fluorescent protein tdTomato (red) in acinar cells. b SMG cells were isolated, dissociated and cultured to allow aggregate formation. c At day 2, aggregates were encapsulated in poly(ethylene glycol) PEG hydrogels. Timeline of cell isolation, aggregate formation, hydrogel encapsulation and analysis of encapsulated aggregates. Scale bars = 50 μm
Acinar cells exhibit low levels of proliferation in vitro
Acinar cells persist in culture for up to 16 days when encapsulated as aggregates in hydrogels (Fig. 2), although numbers decrease with time (Shubin et al. 2015). The percentage of acinar cells within encapsulated SMG aggregates was assessed by counting the tdTomato+ cells using Visiopharm image quantification over the 16-day culture period. As previously reported, after encapsulation at day 2, approximately 90% of cells were of acinar lineage (Online Resource S1c, f). However, the percentage of acinar cells steadily decreased to approximately 35% of the total population by day 9 (Online Resource S1d, f) and remained low through day 16 (Online Resource S1e, f).

Encapsulated aggregates incorporate EdU. Encapsulated SMG aggregates from Mist1CreERT2;R26tdTomato mice were incubated with EdU at day 5 (a), day 12 (b), or day 16 post-isolation (c). tdTomato fluorescence (red) labels Mist1-positive acinar cells. Nuclei were stained with Hoechst 33342. Arrowheads mark acinar cells positive for EdU. The percentage of EdU-positive cells co-localized with tdTomato (Mist1+) compared with EdU-positive cells of non-acinar cell lineage (Mist1−) was quantified at days 5, 12 and 16 (d). Statistical analysis was performed using 2-way ANOVA with Sidak’s multiple comparisons. No significant differences were observed between days 5, 12 and 16. Scale bars = 50 μm. N = 3
Cell proliferation within encapsulated SMG aggregates was assessed through EdU labeling to investigate if expansion of non-acinar cells contributes to the reduction in overall percentage of acinar cells. Encapsulated aggregates were treated with EdU at day 5, 12, or 16 of in vitro culture (Fig. 2a–c). Quantification of EdU+ cells within SMG aggregates showed that most in vitro proliferation occurs in non-acinar cells, although acinar lineage (Mist1+) cells maintain a limited ability to proliferate (Fig. 2d).
Acinar cells downregulate cell-specific markers in vitro after encapsulation in hydrogels
We first examined the fate of acinar cells within SMG aggregates by measuring the expression of several acinar cell-specific markers during aggregate formation. In the intact SMG, MIST1 is expressed in differentiated acinar cells (Pin et al. 2000). The water channel, aquaporin 5 (AQP5), is localized at the luminal surface of acinar cells and the sodium-potassium-chloride channel (NKCC1) is localized on all acinar cell basal membranes and a small subset of duct cells (Arany et al. 2011). Within SMG aggregates, expression of Aqp5 and Mist1 was markedly reduced after 48 h of culture (day 2), while Nkcc1 expression was not significantly changed (Fig. 3). Thus, even during the initial stage of suspension culture, primary acinar cells downregulate several key acinar cell genes.

Primary acinar cells downregulate key cell-specific markers. Relative gene expression of salivary gland acinar cell markers Mist1, Aqp5 and Nkcc1 in SMG primary gland isolate (day 0) and in SMG aggregates at 48 h post-isolation (day 2) was measured using quantitative RT-PCR and normalized to ribosomal protein LE32. N = 6, ± standard deviation, *p < 0.05
To investigate the fate of acinar cells following hydrogel encapsulation, co-expression of tdTomato with AQP5 and NKCC1 was analyzed using immunofluorescence staining (Fig. 4). Two days after cell isolation, a lower proportion (80%) of acinar lineage cells within SMG aggregates expressed NKCC1 compared with native SMG (94%) (Fig. 4b, Online Resource S2). By day 16, the SMG aggregates had developed interior lumens lined by NKCC1+ cells (Fig. 4c), as previously reported (Shubin et al. 2017). Although there was an overlap between tdTomato and NKCC1 in the aggregates, ~ 50% of tdTomato+ acinar lineage cells did not co-stain for NKCC1 (Fig. 4c, arrowhead; Online Resource S2). Notably, by day 16, many NKCC1+ cells were of non-acinar cell origin (Fig. 4c, arrows). These may represent expanded NKCC1+ cells from the ducts.

Expression of NKCC1 and AQP5 is dysregulated in SMG aggregates encapsulated within hydrogels. Immunofluorescence staining on frozen sections of Mist1CreERT2;R26tdTomato SMG with antibodies to NKCC1 (a) and AQP5 (d) shows co-localization with tdTomato-positive acinar cells in vivo. SMG aggregates at day 2 post-isolation stained with antibodies to NKCC1 (b) and AQP5 (e). SMG aggregates at day 16 post-isolation stained with antibodies to NKCC1 (c) and AQP5 (f). Arrows indicate NKCC1-positive cells and AQP5-positive cells that are of non-acinar cell lineage at day 16 (c, f). Arrowheads indicate cells from acinar lineage that are negative for NKCC1, or for AQP5. Nuclei are labeled with DAPI. Scale bars = 50 μm (a, d) or 20 μm (b, c, e, f)
Consistent with the gene expression data, there was also a decrease in AQP5 levels in SMG aggregates compared with native SMG tissue by day 16 post-encapsulation (Fig. 4d–f, Online Resource S2). Furthermore, AQP5+ cells did not co-localize with tdTomato (Fig. 4f, arrows) and ~ 78% of acinar lineage cells did not express AQP5 (Fig. 4f, arrowheads; Online Resource S2). Thus, expression of both NKCC1 and AQP5 is dysregulated in primary SMG aggregates encapsulated in PEG hydrogels.
Acinar cells activate expression of duct cell-specific markers
Acinar-to-ductal metaplasia is a phenomenon observed in primary cultures of pancreatic acinar cells (Means et al. 2005) and could explain the loss of key phenotypic traits by salivary gland acinar cells encapsulated within PEG hydrogels. In particular, acinar-to-ductal metaplasia is associated with loss of Mist1 expression (Karki et al. 2015; Means et al. 2005; Zhu et al. 2004). To determine whether acinar-to-ductal metaplasia occurs during primary culture of salivary gland cells, co-expression of tdTomato with duct cell markers K5, K7 and K19 was assessed. In vivo, K5, K7 and K19 are expressed in duct cells of the SMG with no detectable expression in acinar cells (Born et al. 1987) (Fig. 5a–d, i–l, Online Resource S3a). Although very low levels of keratin expression were found in freshly encapsulated aggregates (Shubin et al. 2017), both pan-keratin and K5, a marker of myoepithelial and duct cells, were increased in SMG aggregates at day 9 (Online Resource S3b, d). At day 16, K19 and K7 were expressed in ~ 52% and 74%, respectively, of acinar lineage cells in encapsulated SMG aggregates (Fig. 5e–h, m–p, arrowheads; Online Resource S4). These data indicate that SMG acinar cells activate expression of duct cell-specific markers when cultured in vitro.

Acinar lineage cells in encapsulated SMG aggregates activate expression of keratins. a–d Section of Mist1CreERT2;R26tdTomato SMG stained with antibodies to tdTomato (RFP) and keratin 19. e–h SMG aggregates in hydrogel at day 16 stained with antibodies to tdTomato (RFP) and keratin 19. Nuclei are stained with DAPI. Individual channels and merged images are shown. A, acinar cells; D, duct. Arrowheads indicate acinar lineage (RFP+) cells co-localized with keratin 19. i–l Section of Mist1CreERT2;R26tdTomato SMG stained with antibodies to tdTomato (RFP) and keratin 7. m–p SMG aggregates in hydrogel at day 16 stained with antibodies to tdTomato (RFP) and keratin 7. Nuclei are stained with DAPI. Individual channels and merged images are shown. A, acinar cells; D, duct. Arrowheads indicate acinar lineage cells co-localized with keratin 7. N = 3, scale bars = 20 μm
Duct ligation injury induces acinar-to-ductal metaplasia in salivary glands in vivo
Conditions of stress or inflammation induce acinar-to-ductal metaplasia in the exocrine pancreas (Strobel et al. 2007). To determine if salivary gland acinar cells exhibit this transition in phenotype in response to in vivo injury, Mist1CreERT2;R26tdTomato mice were used to follow acinar cell fate after duct ligation. The ligation of the main SMG excretory duct in vivo results in atrophy and loss of acinar cells, which are regenerated if the ligation is removed (Tamarin, 1971). Following tamoxifen gavage to activate expression of tdTomato in acinar cells, unilateral duct ligation was performed on Mist1CreERT2;R26tdTomato mice. After 14 days of duct ligation, immunofluorescence staining of SMG sections showed tdTomato-positive acinar lineage cells organized into duct-like structures and co-localized with the duct cell markers K19 (Fig. 6e–h; Online Resource S6; 62%) and K7 (Fig. 6q–t; Online Resource S6; 70%). This co-localization was observed as early as 9 days after duct ligation (Online Resource S5). These data demonstrate that under conditions of stress in vivo, salivary gland acinar cells undergo a phenotypic change to express duct cell markers. After 2 weeks, the ligation was removed and the glands were allowed to regenerate. Notably, after 2 weeks of regeneration, fewer acinar lineage cells co-localized with the ductal cell markers K7 and K19 (Fig. 6i–l,u–x), suggesting that cells undergoing acinar-to-ductal metaplasia may either revert to the acinar phenotype or undergo apoptosis.

Acinar lineage cells activate expression of keratins in vivo after duct ligation. a–d Section of Mist1CreERT2;R26tdTomato SMG control. e–hMist1CreERT2;R26tdTomato SMG after duct ligation. i–lMist1CreERT2;R26tdTomato SMG at 14 days after ligation removal and regeneration stained for tdTomato and keratin 19. Nuclei are stained with DAPI. Individual channels and merged images are shown. A, acinar cells; D, duct. Arrowheads indicate acinar lineage cells co-localized with keratin 19. m–p Section of Mist1CreERT2;R26tdTomato SMG control. q–tMist1CreERT2;R26tdTomato SMG after duct ligation. u–xMist1CreERT2;R26tdTomato SMG at 14 days after ligation removal and regeneration, stained for tdTomato and keratin 7. Nuclei are stained with DAPI. Individual channels and merged images are shown. A, acinar cells; D, duct. Arrowheads indicate acinar lineage cells co-localized with keratin 7. N = 3, scale bars = 20 μm
Discussion
Continued proliferation of differentiated cells plays a major role in maintaining salivary gland acinar cell populations in vivo (Aure et al. 2015b; Emmerson et al. 2018). This suggests that regenerative strategies for the salivary gland could be based on the use of acinar cells for cell replacement. However, it is well established that salivary gland acinar cells quickly lose their secretory phenotype in vitro (Kanamura and Barka, 1975), so that the isolation and expansion of cells for therapeutic use is a limiting factor. Dissociated SMG acinar cells undergo loss of polarization, changes in gene expression and decreased secretion (Quissell et al. 1994, 1986; Redman et al. 1988). Recent evidence shows that specific culture conditions may retain the acinar cell secretory phenotype in vitro (Jang et al. 2015). Culture of SMG aggregates in hydrogels also holds promise as a means to maintain regenerative cells in vitro, as well as to enable cell transplantation for tissue engineering approaches. We and others have reported that in PEG hydrogels, Matrigel, or other natural hydrogels, primary salivary gland cells form aggregates with polarized epithelial layers and increase expression of duct cell markers, although expression of acinar cell markers is decreased (McCall et al. 2013; Pradhan et al. 2010; Pradhan-Bhatt et al. 2013; Shubin et al. 2015, 2017). The in vitro loss of key acinar cell-specific markers in previous studies precluded the investigation of acinar cell fate. In this work, genetic labeling was used to track acinar cell survival and fate after hydrogel encapsulation.
The MIST1 transcription factor is required for establishing the secretory phenotype of pancreatic acinar cells (Johnson et al. 2004; Lo et al. 2017; Pin et al. 2000, 2001). Loss of MIST1 results in cellular disorganization, changes in polarity and loss of secretory function (Johnson et al. 2004; Zhu et al. 2004). These changes are also observed with in vitro culture of pancreatic acinar cells (De Lisle and Logsdon, 1990; Githens et al. 1994; Rooman et al. 2000; Sphyris et al. 2005). Upon initial formation in vitro, SMG aggregates comprise both acinar and duct cells, with the majority cells of acinar origin. By day 9 in culture, the percent of acinar cells is decreased to ~ 35% and remains at this level up to 16 days post-isolation. Although hydrogel-encapsulated SMG aggregates exhibited lumen formation and apical-basolateral cellular organization (Shubin et al. 2017), gene expression analysis revealed that acinar cells within the aggregates rapidly decreased expression of Mist1 and Aqp5. These changes in gene and protein expression are consistent with the loss of the acinar cell phenotype. The use of MMP-degradable hydrogels partially recovered expression of Aqp5 and Nkcc1 in encapsulated SMG aggregates, but did not prevent decreased Mist1 expression during the 2-week culture period (Shubin et al. 2017).
In response to in vivo stress or injury, such as acute pancreatitis (Guerra et al. 2007), pancreatic acinar cells acquire phenotypic characteristics of duct cells, a process known as acinar-to-ductal metaplasia (Jensen et al. 2005; Means et al. 2005; Pan et al. 2013). Down regulation of Mist1 expression is closely associated with this process and MIST1 inhibition in pancreatic acinar cells also leads to activation of duct-specific genes such as K19 (Zhu et al. 2004). Lineage tracing showed that Mist1-expressing acinar cells in the SMG aggregates activated expression of the duct-specific keratins K7 and K19. Consistent with this, in vivo injury induced by duct ligation also led to activated expression of keratins in lineage-traced acinar cells. Together, these data indicate that salivary gland acinar cells respond to stress or in vivo injury by undergoing acinar-to-ductal metaplasia.
This work reveals evidence of cellular plasticity in the salivary gland in response to environmental cues. Silencing of MIST1 is required for survival of pancreatic acinar cells subjected to stress (Karki et al. 2015). Transient downregulation of MIST1 in pancreatic acinar cells shuts down the secretory machinery and permits cell proliferation. Once the stress is removed, the cells regain the secretory phenotype (Karki et al. 2015). This suggests that downregulation of Mist1 expression in SMG aggregates may be necessary for maintaining cell viability in vitro. The activation of K7 and K19 expression in acinar lineage cells following duct ligation shows that acinar-to-ductal metaplasia occurs in vivo. After the salivary gland has recovered, some acinar lineage cells are still detected in the ducts (arrowheads, Figs. 6l, x), suggesting that not all revert to secretory cells. Expression of Nkcc1 and Aqp5 by cells of non-acinar origin in encapsulated SMG aggregates may be due to expansion of NKCC1+ duct cells or AQP5+ intercalated duct cells, but could alternatively suggest that duct and myoepithelial cell types also exhibit cell plasticity (Tata et al. 2018; Weng et al. 2018).
Previous work in our laboratory (Shubin et al. 2017) and others (Cantara et al. 2012; Kwon and Larsen 2015; McCall et al. 2013; Nelson and Larsen, 2015; Pradhan et al. 2010; Pradhan-Bhatt et al. 2013; Soscia et al. 2013) demonstrated that modification of extracellular microenvironments can have a major effect on salivary gland cell phenotype. The in vitro expansion and culture of primary salivary gland cells using innovative biomaterial designs may promote the acinar cell phenotype. This work reveals previously uncharacterized responses of salivary gland acinar cells to primary culture and injury. The long-term viability and proliferation of acinar cells within encapsulated SMG aggregates has potential significance for the development of regenerative therapies. Further investigation will be necessary to determine if, under finely tuned conditions, in vitro culture of whole gland isolates, including acinar cells, may provide an expanded cell population capable of regeneration.
References
Anthis NJ, Clore GM (2013) Sequence-specific determination of protein and peptide concentrations by absorbance at 205 nm. Protein Sci 22:851–858
Arany S, Catalan MA, Roztocil E, Ovitt CE (2011) Ascl3 knockout and cell ablation models reveal complexity of salivary gland maintenance and regeneration. Dev Biol 353:186–193
Aure MH, Arany S, Ovitt CE (2015a) Salivary glands: stem cells, self-duplication, or both? J Dent Res 94:1502–1507
Aure MH, Konieczny SF, Ovitt CE (2015b) Salivary gland homeostasis is maintained through acinar cell self-duplication. Dev Cell 33:231–237
Born IA, Schwechheimer K, Maier H, Otto HF (1987) Cytokeratin expression in normal salivary glands and in cystadenolymphomas demonstrated by monoclonal antibodies against selective cytokeratin polypeptides. Virchows Arch A Pathol Anat Histopathol 411:583–589
Cantara SI, Soscia DA, Sequeira SJ, Jean-Gilles RP, Castracane J, Larsen M (2012) Selective functionalization of nanofiber scaffolds to regulate salivary gland epithelial cell proliferation and polarity. Biomaterials 33:8372–8382
De Lisle RC, Logsdon CD (1990) Pancreatic acinar cells in culture: expression of acinar and ductal antigens in a growth-related manner. Eur J Cell Biol 51:64–75
Desai BM, Oliver-Krasinski J, De Leon DD, Farzad C, Hong N, Leach SD, Stoffers DA (2007) Preexisting pancreatic acinar cells contribute to acinar cell, but not islet beta cell, regeneration. J Clin Invest 117:971–977
Emmerson E, May AJ, Berthoin L, Cruz-Pacheco N, Nathan S, Mattingly AJ, Chang JL, Ryan WR, Tward AD, Knox SM (2018) Salivary glands regenerate after radiation injury through SOX2-mediated secretory cell replacement. EMBO Mol Med 10:e8051
Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS (2009) Photoinitiated polymerization of PEG-diacrylate with lithium phenyl-2,4,6-trimethylbenzoylphosphinate: polymerization rate and cytocompatibility. Biomaterials 30:6702–6707
Githens S, Schexnayder JA, Moses RL, Denning GM, Smith JJ, Frazier ML (1994) Mouse pancreatic acinar/ductular tissue gives rise to epithelial cultures that are morphologically, biochemically and functionally indistinguishable from interlobular duct cell cultures. In Vitro Cell Dev Biol Anim 30A:622–635
Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP, Barbacid M (2007) Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11:291–302
Jang SI, Ong HL, Gallo A, Liu X, Illei G, Alevizos I (2015) Establishment of functional acinar-like cultures from human salivary glands. J Dent Res 94:304–311
Jensen JN, Cameron E, Garay MV, Starkey TW, Gianani R, Jensen J (2005) Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology 128:728–741
Johnson CL, Kowalik AS, Rajakumar N, Pin CL (2004) Mist1 is necessary for the establishment of granule organization in serous exocrine cells of the gastrointestinal tract. Mech Dev 121:261–272
Kanamura S, Barka T (1975) Short term culture of dissociated rat submandibular gland cells. Lab Investig 32:366–372
Karki A, Humphrey SE, Steele RE, Hess DA, Taparowsky EJ, Konieczny SF (2015) Silencing Mist1 gene expression is essential for recovery from acute pancreatitis. PLoS One 10:e0145724
Kwon HR, Larsen M (2015) The contribution of specific cell subpopulations to submandibular salivary gland branching morphogenesis. Curr Opin Genet Dev 32:47–54
Lo HG, Jin RU, Sibbel G, Liu D, Karki A, Joens MS, Madison BB, Zhang B, Blanc V, Fitzpatrick JA, Davidson NO, Konieczny SF, Mills JC (2017) A single transcription factor is sufficient to induce and maintain secretory cell architecture. Genes Dev 31:154–171
McCall AD, Nelson JW, Leigh NJ, Duffey ME, Lei P, Andreadis ST, Baker OJ (2013) Growth factors polymerized within fibrin hydrogel promote amylase production in parotid cells. Tissue Eng A 19:2215–2225
Means AL, Meszoely IM, Suzuki K, Miyamoto Y, Rustgi AK, Coffey RJ Jr, Wright CV, Stoffers DA, Leach SD (2005) Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development 132:3767–3776
Nelson DA, Larsen M (2015) Heterotypic control of basement membrane dynamics during branching morphogenesis. Dev Biol 401:103–109
Pan FC, Bankaitis ED, Boyer D, Xu X, Van de Casteele M, Magnuson MA, Heimberg H, Wright CV (2013) Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development 140:751–764
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45
Pin CL, Bonvissuto AC, Konieczny SF (2000) Mist1 expression is a common link among serous exocrine cells exhibiting regulated exocytosis. Anat Rec 259:157–167
Pin CL, Rukstalis JM, Johnson C, Konieczny SF (2001) The bHLH transcription factor Mist1 is required to maintain exocrine pancreas cell organization and acinar cell identity. J Cell Biol 155:519–530
Pradhan S, Liu C, Zhang C, Jia X, Farach-Carson MC, Witt RL (2010) Lumen formation in three-dimensional cultures of salivary acinar cells. Otolaryngol Head Neck Surg 142:191–195
Pradhan-Bhatt S, Harrington DA, Duncan RL, Jia X, Witt RL, Farach-Carson MC (2013) Implantable three-dimensional salivary spheroid assemblies demonstrate fluid and protein secretory responses to neurotransmitters. Tissue Eng A 19:1610–1620
Quissell DO, Redman RS, Barzen KA, McNutt RL (1994) Effects of oxygen, insulin and glucagon concentrations on rat submandibular acini in serum-free primary culture. In Vitro Cell Dev Biol Anim 30A:833–842
Quissell DO, Redman RS, Mark MR (1986) Short-term primary culture of acinar-intercalated duct complexes from rat submandibular glands. In Vitro Cell Dev Biol 22:469–480
Redman RS, Quissell DO, Barzen KA (1988) Effects of dexamethasone, epidermal growth factor and retinoic acid on rat submandibular acinar-intercalated duct complexes in primary culture. In Vitro Cell Dev Biol 24:734–742
Rooman I, Heremans Y, Heimberg H, Bouwens L (2000) Modulation of rat pancreatic acinoductal transdifferentiation and expression of PDX-1 in vitro. Diabetologia 43:907–914
Ruan JL, Tulloch NL, Muskheli V, Genova EE, Mariner PD, Anseth KS, Murry CE (2013) An improved cryosection method for polyethylene glycol hydrogels used in tissue engineering. Tissue Eng Part C Methods 19:794–801
Rugel-Stahl A, Elliott ME, Ovitt CE (2012) Ascl3 marks adult progenitor cells of the mouse salivary gland. Stem Cell Res 8:379–387
Shubin AD, Felong TJ, Graunke D, Ovitt CE, Benoit DS (2015) Development of poly(ethylene glycol) hydrogels for salivary gland tissue engineering applications. Tissue Eng A 21:1733–1751
Shubin AD, Felong TJ, Schutrum BE, Joe DSL, Ovitt CE, Benoit DSW (2017) Encapsulation of primary salivary gland cells in enzymatically degradable poly(ethylene glycol) hydrogels promotes acinar cell characteristics. Acta Biomater 50:437–449
Soscia DA, Sequeira SJ, Schramm RA, Jayarathanam K, Cantara SI, Larsen M, Castracane J (2013) Salivary gland cell differentiation and organization on micropatterned PLGA nanofiber craters. Biomaterials 34:6773–6784
Sphyris N, Logsdon CD, Harrison DJ (2005) Improved retention of zymogen granules in cultured murine pancreatic acinar cells and induction of acinar-ductal transdifferentiation in vitro. Pancreas 30:148–157
Strobel O, Dor Y, Alsina J, Stirman A, Lauwers G, Trainor A, Castillo CF, Warshaw AL, Thayer SP (2007) In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology 133:1999–2009
Tamarin A (1971) Submaxillary gland recovery from obstruction. I Overall changes and electron microscopic alterations of granular duct cells J Ultrastruct Res 34:276–287
Tata A, Kobayashi Y, Chow RD, Tran J, Desai A, Massri AJ, McCord TJ, Gunn MD, Tata PR (2018) Myoepithelial cells of submucosal glands can function as reserve stem cells to regenerate airways after injury. Cell Stem Cell 22(668–683):e666
Weng PL, Aure MH, Maruyama T, Ovitt CE (2018) Limited regeneration of adult salivary glands after severe injury involves cellular plasticity. Cell Rep 24(1464–1470):e1463
Zhu L, Tran T, Rukstalis JM, Sun P, Damsz B, Konieczny SF (2004) Inhibition of Mist1 homodimer formation induces pancreatic acinar-to-ductal metaplasia. Mol Cell Biol 24:2673–2681
Acknowledgments
The authors thank Linda Callahan and the University of Rochester Medical Center Confocal and Conventional Microscopy Core for the assistance in imaging. They would also like to thank the University of Rochester Center for Musculoskeletal Research for the use of facilities and Visiopharm analysis software to quantitate the percentage of cells expressing tdTomato (VisioPharm funding provided by 1S10 RR027340-01, P30 AR061307, P50AR054041).
Funding
Research reported in this publication was supported by the National Institute of Dental and Craniofacial Research, the National Center for Advancing Translational Sciences, the National Cancer Institute and the National Institute for General Medical Sciences of the National Institutes of Health under Award Numbers R01DE022949, R56DE025098, UG3DE027695, F30CA183320 (to ADS) and T32GM7356 (to ADS).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. This work was approved by the University Committee on Animal Resources at the University of Rochester (protocol no. 101559/2006-023; approved 8/31/2017 to 8/31/2020).
Informed consent
Not applicable.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 15299 kb)
Rights and permissions
About this article

Cite this article
Shubin, A.D., Sharipol, A., Felong, T.J. et al. Stress or injury induces cellular plasticity in salivary gland acinar cells. Cell Tissue Res 380, 487–497 (2020). https://doi.org/10.1007/s00441-019-03157-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-019-03157-w